Received Date:
26 April 2019 Accepted Date:
26 August 2019 Available Online:
01 November 2019
Abstract:
Three-dimensional porous dendritic Pt films (3D PDPFs) were prepared by cathodic deposition utilizing hydrogen bubble dynamic template at room temperature. The deposition conditions including deposition time, electrolyte and substrate were investigated in detail. The deposited PDPFs were characterized by scanning electron microscopy (SEM) and X-ray diffraction (XRD). The prepared 3D PDPFs have high catalytic activity for methanol electrocatalytic oxidation. After adsorption of pyridine, the material has surface-enhanced Raman scattering (SERS) signals, and it exhibits super hydrophobic performance after modifying a single layer of n-dodecanethiol. The present method provides a new simple strategy toward the fabrication of 3D PDPFs with multiform applications.
High-surface area Pt material with porous structures has many applications because of the extraordinary physical and chemical properties[1~5]. To data, templating[6, 7] and dealloying[8~10] are two most popular methods while they involve multistep to get the ultimate porous structures. Recently, hydro-gen bubbles have been utilized as a dynamic tem-plate in electrodeposition to produce self-supported 3D porous metals under highly cathodic polarization without need of template elimination. Although the hydrogen bubble dynamic template has been developed in electrodeposition of self-supported 3D foam films like Pd[11], Au[12, 13], Ag[14], Sn[15], Cu[16], Ni[17, 18], CuSn[19], PdPt[20], AuPt[21] and CuPd[22], the direct synthesis of 3D porous Pt films with it is still challenging. Thus, an indirect route has been proposed recently including three steps[23]: (1) prepare a 3D porous Cu film first by electro-deposition using the hydrogen bubble template, (2) then immerse the porous Cu film in an aqueous solution of H2PtCl6 solution for 4 h to replace Cu with Pt using the porous Cu as a 3D template, and (3) dissolve the remaining Cu selectively through electrochemical dealloying.
In this study, we report a straightforward approach of direct electrodeposition of 3D porous Pt films utilizing the hydrogen bubble template. The experimental conditions for the deposition were investigated. Some potential applications of as-prepared 3D porous dendritic Pt films (PDPFs) were preliminarily demonstrated including the surface-enhanced Raman scattering (SERS), electrocatalytic oxidation of methanol and wetting property.
1.
Experimental
1.1
Reagents
Hydrogen hexachloroplatinate (Ⅳ) hexahy-drate (H2PtCl6·6H2O), D-(+)-Glucose (C6H12O6·H2O), Na2HPO4·12H2O, NaH2PO4· 2H2O, n-dodecylmercaptan were obtained from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). L-ascorbic acid (AA, 99%) was purchased from Guangdong Xilong Chemical Co. Ltd (Guangzhou, China). Uricacid (UA) was provided by Sigma. Sulfuric acid and methanol were purchased from the Factory of Hunan Normal University. All the chemicals are of analytical grade and are used as received. Milli-Q water with a resistivity of greater than 18.3 MΩ·cm was used in the preparation of aqueous solutions.
1.2
Electrodeposition of 3D porous Pt films using the hydrogen dynamic template
Electrochemical experiments were performed on a CHI 660C electrochemical workstation (Chenhua Instruments, Shanghai, China). A platinum disk (1mm diameter, purity 99.99%), a platinum foil (geometric area of 1cm2), and a saturated mercurous sulfate electrode (SMSE) were employed as the working, counter, and reference electrodes, respectively. The working electrode was polished with 2000 grit carbimet paper before use, followed by rinsing in Millipore water under ultrasonic waves. Then the electrode was electrochemically pretreated by cycling the potential between -0.72 and +0.8 V in 0.5 mol/L H2SO4 at a scan rate of 100 mV/s until a stable voltammogram was obtained. Typically, a 3D porous Pt film was fabricated by electrodeposition on the Pt electrode under constant potential of -4V for 300 s in a solution of 2 mmol/L H2PtCl6+ 2 mol/L H2SO4 at room temperature (~25 ℃). The porous film was used in subsequent property measurements unless otherwise stated. For comparison, deposition time was also investigated by using glass carbon disk (3 mm diameter) substrate and 0.2 mol/L NH4Cl supporting electrolyte.
1.3
SERS measurement
SERS tests were preformed with a Renishaw RM1000 confocal Raman microspectrometer (Glouc-estershire, UK). A detailed description of the Raman measurements with a spectroelectrochemical cell can be found elsewhere[24]. Potential-dependent SERS spectra on the 3D PDPFs were tested in 0.01 mol/L pyridine + 0.1 mol/L KCl, the exciting wavelength was 633 nm, and the collecting time was 10 s.
1.4
Electrooxidation of methanol
The deposited Pt catalysts or bulk Pt electrode were firstly treated by cyclic voltammetry between -0.72 and 0.8 V at 100 mV/s in 0.5 mol/L H2SO4 solution until a steady cyclic voltammogram (CV) was obtained. The electrochemical active surface areas (ECSAs) are calculated according to the integrated charge of hydrogen adsorption at the 3D PDPFs or bulk Pt electrode in 0.5 mol/L H2SO4 with a converting factor of 210 μC·cm-2[26]. The electrocatalytic activity toward the electrooxidation of methanol was evaluated in a solution of 0.5 mol/L H2SO4+1 mol/L CH3OH. And the CVs were recorded in the potential ranges between -0.72 and 0.8 V for the oxidation of methanol, at a scan rate of 50 mV/s. The oxidation current has been normalized to ECSAs of 3D PDPFs or bulk Pt electrode for comparison.
1.5
Wettability tests
Wetting tests were preformed on the surfaces of smooth Pt and 3D PDPFs with and without assembled monolayer of thiol, respectively, by placing a drop of water and taking pictures with a contact angle measuring instrument (JC2000C1). The smooth Pt electrode and 3D PDPFs were cleaned by CV (-0.72 to 0.8 V) in 0.5 mol/L H2SO4 before the tests. The self-assembled thiol monolayer on the surfaces of the smooth Pt and the 3D PDPFs was formed by immersing them overnight in the ethanol solution of n-dodecanethiol (5 mmol/L).
1.6
Morphology and XRD patterns of the 3D PDPFs
Scanning electron microscopy (SEM) images were obtained with a JEOL JSM-6360 scanning electron microscope operating at 25 kV. X-ray diffraction (XRD) patterns were obtained with a Dmax Rapid IIR diffractometer using CuKα (0.1542 nm) radiation, and the exposure time was 15 min.
2.
Results and Discussion
2.1
Characterization of the electrodep-osited porous Pt films
Fig. 1 shows the morphologies of 3D porous Pt films with different deposition time. Apparently, the film thickness and the pore size increased with the increase of deposition time. In 60 s (Fig. 1. a1~a3), only scattered dendrite Pt appeared. When the deposition time was prolonged to 120 s, the film reached a certain thickness and H2 bubbles began to guide the formation of macropores with an average diameter of 3 mm (Fig. 1. b1~b2). The pore size reached to 5~8 mm (Fig. 1. c1~c2), and the dendritic wall became thicker with interconnected nanopores. The growth mechanism of the 3D nanostructures is schematically shown in earlier report[15]. Such porous films with dendrite morp-hology not only have high surface areas but also facilitate fast mass transfer of gas and liquid, which are beneficial for the electrocatalytic reactions.
Figure 1.
SEM images with different enlargement scales for the 3D Pt porous films prepared in a solution of 2 mmol/L H2PtCl6+2 mol/L H2SO4 by electrodepositioned along with H2 evolution at constant potential of -4 V treatment for different periods of time: (a1~a3) 60 s, (b1~b3) 120s, (c1~c3) 300 s at room temperature, substrate: bulk Pt electrode
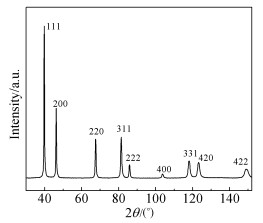
Fig. 2 shows the XRD patterns of the deposited porous Pt films. The peaks at 39.78°, 46.23°, 67.53°, 81.39°, 85.87°, 103.73°, 118.04°, 123.14° and 148.77° are assigned to Pt (111), (200), (220), (311), (222), (400), (331), (420) and (422), respectively, according to JCPDS 04-0802 of cubic FCC phase Pt.
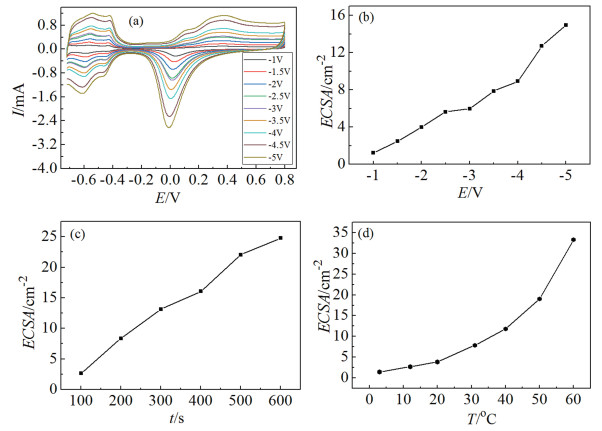
The electrochemically active surface area of the porous Pt was measured in 0.5 mol/L H2SO4 solution, as described in the experimental section. A series of typical CVs are shown in Fig. 3(a) for the 3D PDPFs deposited at different deposition potentials for 200 s at room temperature. Features associated with hydrogen adsorption and desorption events, as well as the reduction of surface oxide, can be seen in the plots. The ECSAs of the porous films depend on the deposition potential, the deposition time, and the bath temperature. We note that, for a given concentration of precursor (2 mmol/L H2PtCl6) and deposition time (200 s) at room temperature, the ECSA increased with the negative shift of the cathodic potentials (Fig. 3 (b)). As can be seen in Fig. 3(c) and Fig. 3(d), ECSAs increased with the increase of deposition time and bath temperature. Both higher temperature and stronger cathodic polarization facilitate fast mass transfer of precursor[26, 27].
Figure 3.
CVs in 0.5 mol/L H2SO4 at a sweep rate of 100 mV/s on the deposited Pt films with a deposition time of 200 s at different deposition potentials at room temperature (a). The dependence of ECSA with (b) deposition potential, (c) deposition time at -4 V at room temperature, and (d) the deposition temperature at -4V for 200 s
In addition, supporting electrolyte and substrate properties also have an effect on the deposition structure of Pt. Replacing 2 mol/L H2SO4 with 0.2 mol/L NH4Cl (in Fig. 4 (a1~a2)), the film thickness of the obtained Pt sphere was about 0.25~0.5 mm, suggesting that a stronger acidic electrolyte was more beneficial to prepare 3D porous Pt films for the hydrogen bubbles formed more easily. When a GC electrode was used as the substrate under the identical deposition conditions, the deposited porous structures of Pt were not as uniform as on the Pt substrate, probably because hydrogen evolution was more favorable on Pt than on the GC substrate.
Figure 4.
SEM images with different enlargement scales for the deposited Pt films prepared at constant potential of -4 V for 300s at room temperature (a1, a2) in a solution of 2 mmol/L H2PtCl6+0.2 mol/L NH4Cl on the substrate of bulk Pt, or (b1, b2) in a solution of 2 mmol/L H2PtCl6+2 mol/L H2SO4 on the substrate of GC
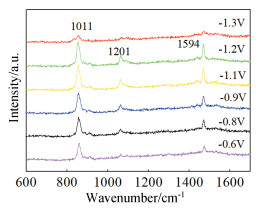
The 3D PDPFs exhibited fairly strong SERS effect as evidenced by the characteristic band for the adsorbed pyridine (Fig. 5). The peaks at 639, 1011, 1209 and 1594 cm-1 are assigned to different ring breathing modes of ν6a, ν1, ν12, and ν8a, respe-ctively[28]. Their intensity and location varied with the applied potential as discribed in the literature[24]. The merit of this substrate for SERS is that it can be repeatedly used for many times as long as cleaning it electrochemically in a H2SO4 solution[24]. We provide here a facile method to prepare effective SERS substrates of Pt for potential applications on mechanism investigation in electrocatalysis.
2.3
Electrocatalytic activity toward methanol oxidation on the deposited 3D PDPFs
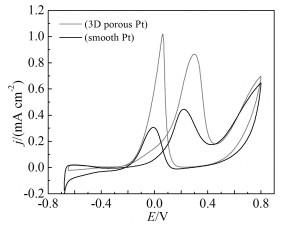
The electrocatalytic activity of the 3D PDPFs toward to the oxidation of methanol was measured in an aqueous solution of 0.5mol/L H2SO4 and 1mol/L methanol between -0.65 V and 0.8 V in comparison with smooth Pt based on the ECSA (Fig. 6). During positive-going potential scan, the peak current density of the 3D PDPFs at 0.29 V was 0.84 mA/cm, about two times as large as on the bulk Pt. The high electrocatalytic activity of the 3D PDPFs electrode toward oxidation of methanol can be attributed to the special porous structure and the nano-dendrite walls. The porous structure allows efficient mass transfer of liquid fuel and alleviates the concentration polarization during reaction, and the nano-dendrites promote the surface reactions.
Figure 6
图 6.
3D PDPF和块状Pt电极在1mol/L CH3OH+0.5mol/L H2SO4溶液中扫速50 mV/s下的CV图
Figure 6.
CVs of on the 3D PDPF and bulk Pt electrode in 1 mol/L CH3OH+0.5 mol/L H2SO4 with a sweep rate of 50mV/s
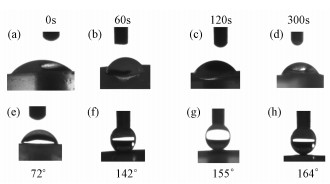
Controlling the wettability of solid surfaces by tailoring surface morphology and surface chemical compositions is an important issue, which attracts the increasing interest from both fundamental and practical perspective[17, 29]. The wettability is usually determined by measuring the contact angle (CA) of a water droplet on a solid surface. When the CA of a water droplet on the surface is larger than 150°, it is called superhydrophobic. Artificial superhydrophobic surfaces are fabricated on the basis of the inspiration of the lotus leaf by employing two kinds of approaches: creating hierarchical structures (micro- and nanostructures) on hydrophobic substrates, or chemically modifying a hierarchical structured surface with a low surface free energy material[30]. As shown in Fig. 7 (a~d), the 3D PDPFs and Pt substrate without decorating n-dodecanethiol monolayer demonstrated the hydrophilicity with a contact angle less than 72°. However, after assembling n-dodecanethiol monolayer, the surface of 3D PDPFs exhibited not only hydrophobicity with a large contact angle of 142° (Fig. 7f) even over 150° (Fig. 7g~h) but also excellent antiadhesion capability since the water droplet can hardly be transferred from the pipet tip to the surface and easily slided off the substrate by even very slight perturbation. Whereas, after being coated with n-dodecanethiol monolayer, the surface of smooth Pt substrate was still hydrophilic. Therefore, the wettability of 3D PDPFs is governed by both the geometrical microstructure and the surface free energy of the surface.
Figure 7.
Photos of water droplets on the surfaces of (a, e) smooth Pt and (b, c, d, f, g, h) 3D PDPFs in the case of (a~d) without and (e~h) with the assembled n-dodecanethiol monolayer
In conclusion, macroporous Pt films with nano-dendrite walls can be directly deposited by utilizing hydrogen bubble template. The morphology depends more or less on the applied potential, deposition time, bath temperature, substrate as well as the supporting electrolyte. Due to the special micro-nano structure, the 3D PDPFs have some significant potential applications in electrocatalysis, surface-enhanced Raman scattering and superhydrophobicity.
H Y Erbil, A L Demirel, Y Avci et al. Science, 2003, 299: 1377~1380. doi: 10.1126/science.1078365
Figure 1
SEM images with different enlargement scales for the 3D Pt porous films prepared in a solution of 2 mmol/L H2PtCl6+2 mol/L H2SO4 by electrodepositioned along with H2 evolution at constant potential of -4 V treatment for different periods of time: (a1~a3) 60 s, (b1~b3) 120s, (c1~c3) 300 s at room temperature, substrate: bulk Pt electrode
Figure 3
CVs in 0.5 mol/L H2SO4 at a sweep rate of 100 mV/s on the deposited Pt films with a deposition time of 200 s at different deposition potentials at room temperature (a). The dependence of ECSA with (b) deposition potential, (c) deposition time at -4 V at room temperature, and (d) the deposition temperature at -4V for 200 s
Figure 4
SEM images with different enlargement scales for the deposited Pt films prepared at constant potential of -4 V for 300s at room temperature (a1, a2) in a solution of 2 mmol/L H2PtCl6+0.2 mol/L NH4Cl on the substrate of bulk Pt, or (b1, b2) in a solution of 2 mmol/L H2PtCl6+2 mol/L H2SO4 on the substrate of GC
Figure 7
Photos of water droplets on the surfaces of (a, e) smooth Pt and (b, c, d, f, g, h) 3D PDPFs in the case of (a~d) without and (e~h) with the assembled n-dodecanethiol monolayer


 下载:
下载:


 下载:
下载:








