使用可见光催化光解水制取氢气始终是能源和环境催化领域的重要挑战之一[1 2 。在过去几十年里, 为提高光催化产氢过程的反应性和选择性, 化学家进行了大量的前期工作, 发现了多种光催化剂, 如TiO2 、敏化纳米结构金属氧化物、多金属氧簇类等[1 15 。其中, 多金属氧酸盐(简称多酸, POM)作为结构多样的廉价无机催化剂, 因其非常宽的吸收光谱几乎覆盖了整个可见光区, 而被作为优良的电子受体和给体, 在催化光解水制氢领域显示了很好的潜在利用价值[5 15 。
早期, Darwent等[12 使用Si取代Keggin型多酸[SiW12 O40 ]4- (简写为{Si}4- )为光催化剂、甲醇为牺牲还原剂, 在λ > 300nm的光源照射下裂解水制取氢气。他们推测, {Si}4- 被光激发后从甲醇获得电子, 生成单电子还原(OER)中间体{Si}5- , 接着两个{Si}5- 和H+ 作用产生H2 。反应过程为:
后期, Keïta等[13 将{Si}4- 电化学沉积在玻碳电极等系列电极材料表面。结果显示电催化产氢效果并未出现较大差异。据此, 他们推测氢气可能产生于{Si}4- 表面。李灿等[14 通过电化学实验发现{Si}4- 发生四电子还原需要较高的外界能量(3. 94eV), 在λ>300nm区域, 主要活性物种应为双电子还原(TER)中间体, {Si}6- 。与Darwent等[12 不同的是, 他们推测OER中间体{Si}5- 在激发光作用下, 与甲醇反应生成{Si}6- 。接下来, {Si}6- 和H+ 作用生成H2 。反应过程为:
王恩波等[15 合成了三乙基胺(TEA)配位的{Si}4- 。他们发现牺牲还原剂TEA与多酸的直接配位, 可以有效促进电子转移, 进而裂解水产生H2 , 催化效率也高于甲醇作为牺牲还原剂。遗憾的是文中并未阐述催化机理。
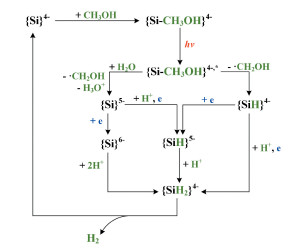
为探索过渡金属类催化剂催化光解水制取H2 的机理, 研究者[16 18 分别计算了系列的钨氧化物、铱、钌和铑配合物催化光解水产氢机理, 发现质子转移和电子转移在整个催化循环中扮演了重要角色, 特别是中间体的质子化对催化反应能垒具有较大影响。Bagno等[19 20 对[W6 O19 ]4+ 、[W10 O32 ]4+ 和[PW12 O40 ]3- 的紫外-可见光谱(UV-Vis)进行了模拟, 发现多酸在激发光作用下发生氧原子到金属, 即配体到金属的电荷转移跃迁(LMCT), 导致端氧原子具有自由基特征。基于前人前期实验和理论研究, 我们推测了{Si}4- 催化光解水产H2 的循环过程(图 1 4- 和甲醇配位生成{SiCH3 OH}4- , 在外界光源刺激下, 跃迁至激发态, 并伴随着甲醇的一个电子转移到多酸表面, 生成OER中间体。接下来, OER中间体从外界得到另一个电子, 生成TER中间体。最后, TER中间体裂解水释放H2 。
图 1
本文通过密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)证实我们所提出的机理, 构建完整的多酸催化光解水产氢反应的势能面, 明确相应的电子和质子转移机理, 找到最有利的反应路径, 特别是明确多酸在催化循环中的角色, 为未来多酸类光催化剂的设计提供理论信息。
1.
计算方法
所有计算均采用Gaussian 16 C. 01程序包[21 。几何优化采用B3LYP杂化泛函[22 24 。对W原子采用赝势基组LanL2DZ[25 , C、H和O原子采用6-31G(d, p)基组[26 28 。所有优化的几何结构都进行了频率计算, 用来确定稳定点和过渡态。使用TD-DFT计算激发态的电子性质[29 30 。使用M06[31 、M08[32 、X3LYP[33 、CAMB3LYP[34 、BP86[35 36 和wb97XD[37 泛函验证模拟的紫外光谱。使用GaussSum 3. 1[38 分析和绘制可见光谱。所有DFT和TD-DFT计算中均采用严格的收敛标准(10-10 au), 并均采用IEF-PCM溶剂模型,H2 O为溶剂[39 。
2.
结果和讨论
2.1
催化剂结构和吸收光谱
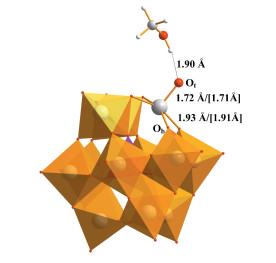
优化的{Si}4- 主要键参数很好重现了XRD结果[12 15 40 , W—Ot 、W—Ob 、Si—O键的最大误差仅为0.02Å。根据前期结果[5 11 , {Si}4- 的主要活性位点为端氧Ot 原子。甲醇水溶液中, 多酸可以与CH3 OH或H2 O配位。计算结果显示, 多酸与CH3 OH或H2 O配位后分别放热3. 6和5. 6kcal· mol- 1 。尽管多酸与水分子配位放热略多, 但{Si}4- + H2 O → {SiH }4- +·OH反应需吸热81. 9kcal· mol- 1 , 远高于甲醇解离所需的62. 5kcal · mol- 1 (表 1 1 ), 这也符合Darwent等[12 和李灿等[14 的实验推测, 即牺牲还原剂主要是甲醇。接下来的计算将采用{Si}4- 与甲醇配位的结构(图 2 4- 的UV-Vis吸收峰位于约257nm[40 , 而催化反应的光源为λ>300nm[12 14 15 。推测实验观测到的UV-Vis吸收峰应来自CH3OH与{Si}4- 配位后发生的电子跃迁。
表 1
图 2
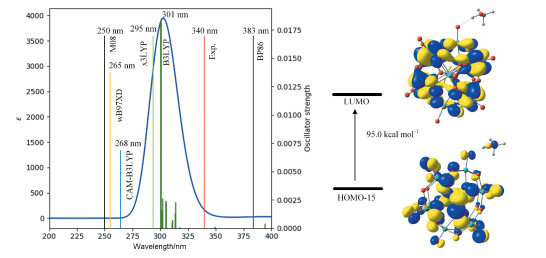
TD-DFT计算结果显示最大吸收峰位于301nm。这也符合文献计算结果[5 8 , 即多酸与溶剂配位后, 紫外吸收峰将发生红移。为了验证计算结果的可靠性, 使用多种泛函对{Si-CH3 OH}4- 的UV-Vis吸收峰进行计算, 结果显示B3LYP模拟的UV-Vis光谱和实验结果最吻合(图 3 2p 电子到Wd 轨道的跃迁, 即配体到金属的LMCT。可以预测缺电子的Ot 原子将具有自由基特征, 促进多酸发生后续的电子和质子转移。
图 3
2.2
光催化机理
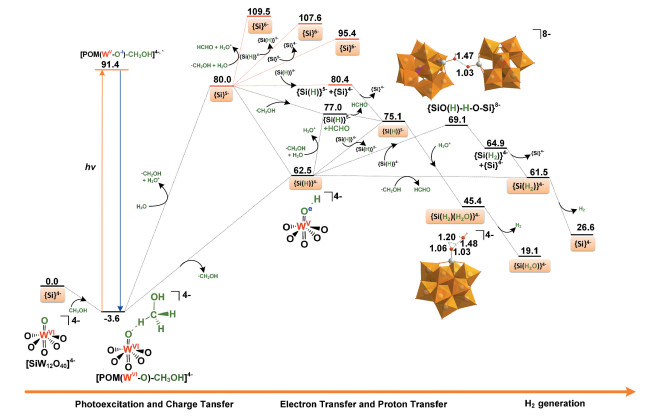
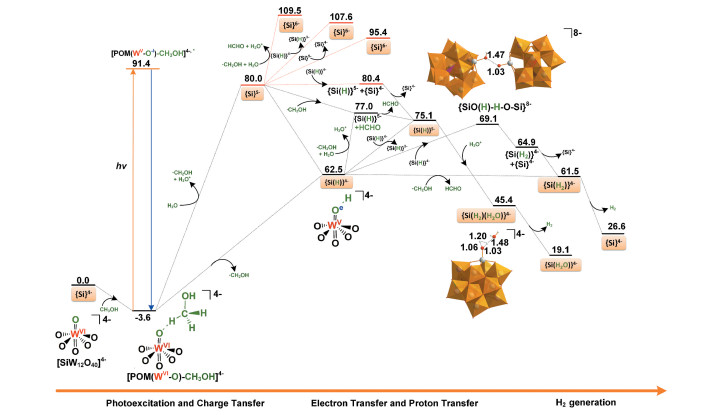
图 4 表 1 4- 催化光解水产氢的反应势能面和相关路径的能量。注意, 此处所有结构均在水溶剂中进行全优化。可以看出, {Si}4- 裂解水制取氢气至少需要克服62. 5kcal· mol-1 的能垒, 室温下不能发生。可见光照射下, {Si}4- 与光波相互作用, 当电子从牺牲还原剂转移到多酸, 就能出现所有步骤均放热的路径。对比多种泛函的计算结果发现, 泛函没有改变势能面吸热或放热的本质。例如, 使用CAM-B3LYP计算的反应1 , 吸热为65. 8kcal· mol- 1 。接下来, 将对光激发后{Si}4- 裂解水制取氢气的三个主要步骤进行详细讨论, 即(1)分子间电荷转移生成OER中间体{Si(H)}4- 或{Si}5- ; (2) OER中间体{Si(H)}4- 或{Si}5- 从外界电子给体获取电子成TER中间体{Si}6- 、{Si(H)}5- 或{Si(H2 )}4- ; (3) TER中间体{Si}6- 、{Si(H)}5- 或{Si(H2 ) }4- 催化裂解水生成H2 , 并重生催化剂。
图 4
2.2.1
电荷转移和OER中间体生成
上面提到{Si}4- 发生LMCT跃迁后, 与甲醇配位的Ot 原子成为缺电子中心, 诱导甲醇的一个电子转移到Ot 原子。下面的计算结果将看到激发态发生电子或质子转移所需能量将超过实验光源的阈值(λ > 300nm)。最有可能的路径应为发生光激发后, 通过能量弛豫回到基态完成后续反应, 这也符合文献[12 14
分别对甲醇的羟基氢原子H(O)和甲基氢原子H(C)的转移进行柔性扫描。计算结果显示, 两种H原子的转移均为无能垒的吸热过程, 分别吸热62. 5kcal· mol- 1 (H-O键断裂, 反应1 )和80. 0kcal· mol- 1 (H-C键断裂, 反应1′ )。因H(O)与多酸Ot 原子形成分子间氢键, 扫描结果显示H (O)原子从甲醇直接转移到多酸Ot 原子, 而离去的H(C)原子与溶剂H2 O分子结合生成H3 O+ , 这导致H(C)原子转移过程的总反应热高于H(O)转移过程17. 5kcal· mol-1 。因此, 研究者[12 14 推测式(2)和(6)的实际过程应为耦合了质子转移的电子转移(反应1 )。NBO计算结果显示生成的中间体{POM-W = OeH-OCeH3 }4- 为具有两个未成对电子的单重态(图 5 - 1 , 导致总能量超过100kcal· mol- 1 , 远高于实验阈值。这也符合实验推测, 即能量弛豫是体系回到基态的最可能方式。总之, 多酸与甲醇配位后, 在光照下发生电子跃迁, 通过能量弛豫回到基态, 发生分子间的电子耦合质子转移, 生成OER中间体{Si(H)}4- 。此处, 因{Si}5- 可通过进一步的质子化生成{Si(H)}4- , 接下来, 我们也将计算{Si}5- 参与的所有路径。
图 5
2.2.2
OER中间体反应性和TER中间体生成
根据李灿等[14 的实验结果, 光催化裂解水产氢是一个双电子还原过程, 这需要OER中间体{Si}5- 或{Si(H)}4- 从外界得到一个电子, 生成TER中间体{Si}6- , {Si(H)}5- 或{Si(H2 ) }4- 。根据溶液中可能的电子给体(CH3 OH, · CH2 OH, {Si}5- 或{Si(H)}4- ), OER中间体主要有三种方式获得电子生成TER中间体。即, (1) {Si}5- 或{Si(H)}4- 从CH3 OH获得电子, 即反应2 , 2′ , 3 或3′ 。计算结果显示这些反应至少需吸热62. 9kcal · mol- 1 。李灿等[14 推测通过光激发可以诱导甲醇转移电子到{Si}5- 或{Si(H)}4- 来产生TER中间体。而使用TD-DFT计算的{Si}5- 垂直激发能为39. 2kcal· mol- 1 (实验值730nm, 40. 6kcal· mol- 1 )10 , {Si(H)}4- 的垂直激发能为37. 1kcal· mol- 1 , 均无法满足反应2 , 2′ , 3 或3′ 所需能量。这需要{Si}5- 或{Si(H)}4- 寻找其他的电子来源产生TER中间体。(2) {Si}5- 或{Si(H)}4- 从甲醇自由基· CH2 OH获得电子, 即反应4 和5 。计算结果显示这两个反应分别吸热29. 5和14. 5 kcal· mol- 1 。而该过程为电子转移耦合质子转移时, 即反应4′ 和5′ , 则变为热力学有利的放热过程, 分别放热3. 0和1. 0 kcal· mol- 1 , 即质子化有利于活性中间体的生成。NBO计算结果显示伴随着质子转移, 电子已从甲醇自由基转移到多酸(图 5 4′ 和5′ 分别生成{Si(H)}5- + HCHO或{Si(H2 ) }4- +HCHO, 这也解释了产物HCHO的来源。(3) {Si}5- 或{Si(H)}4- 从自身获得电子, 即反应6 , 7 , 7′ , 8 或8′ 。DFT计算结果显示只有7′ 和8 需要克服的反应能垒最小。此处, 虽然反应7′仅需吸热0. 4kcal· mol-1 , 但因{Si}5- 的生成在能量上不利, 我们不再对反应7′的详细过程进行计算。对于反应8 , 即2{Si(H)}4- → {Si}4- + {Si (H2 ) }4- , 吸热2. 4kcal· mol- 1, 这在室温下也不难实现, 而且后续反应也均为热力学上有利的放热过程。计算结果显示, 两个{Si(H)}4- 先发生二聚生成{Si-O1 (H1 ) -H2 -O2 -Si}8- , 吸热6. 6kcal· mol- 1 。进一步通过柔性扫描H2 -O2 键, 结果显示该H2 向O1 的转移过程为一个无能垒过程, 放热约4. 3kcal· mol-1 。NBO计算表明其中一个多酸的电子也伴随着H2 转移到另一个多酸的O1 原子。总之, OER中间体{Si}5- 或{Si(H)}4- 通过甲醇自由基和自身发生电子转移反应生成TER中间体{Si}6- 在能量上是不利的(反应2′ , 4 , 6 和7 ), 而发生电子耦合质子转移生成{Si(H)}5- 和{Si(H2 ) }4- (反应4′ , 5′ 和8 )在能量上是有利的。
2.2.3
TER中间体反应性和H2生成
DFT计算结果显示尽管TER中间体{Si}6- 释放H2 均为热力学上有利的放热过程(反应9~ 11 ), 但上面提到{Si }6- 的生成需要吸热至少95. 4kcal· mol- 1 (反应1′ 和6 ), 在能量上是不利的, 需要更强的外界光源, 并不符合实验研究的目标, 即使用可见光源, 这就需要由TER中间体{Si (H2 ) }4- 或{Si(H)}5- 催化裂解水产生H2 。
对于{Si (H2 ) }4- , 我们多次试图寻找{Si (H2 ) }4- → {Si}4- + H2 (反应12 )的过渡态, 结果显示H2 的离去是一个无能垒放热过程, 放热34. 9kcal· mol- 1 。这也符合Fortage等[18 的计算结果, 即两个H原子与一个反应中心配位后, H2 的产生是一个快速且无能垒的放热过程。注意, 生成{Si(H2 ) }4- 的反应5 和8 均为放热过程, 因此, 一旦OER中间体{Si(H)}4- 生成, 接下来的反应5 、8 和12 均为热力学上有利的放热过程(图 4
相比{Si(H2 ) }4- , {Si(H)}5- 催化裂解水产H2 还需要一次质子化, 即与H3 O+ 配位, 形成水分子配位的双氢中间体{Si (H2 ) (H2 O) }4- (反应13 )。柔性扫描结果显示反应13 是一个无能垒放热过程, 放热29. 7kcal· mol- 1 。接下来, 与{Si (H2 ) }4- 释放H2 过程类似, {Si(H2 ) (H2 O) }4- 释放H2也是一个无能垒放热过程, 放热26. 3kcal· mol- 1 (反应14 ), 并重生催化剂{Si(H2 O)2 }4- , 完成一个催化循环。
2.3
对比实验
Darwent等[12 推测光激发后的{Si}4-* 与甲醇发生电子转移, 生成单电子还原中间体{Si}5- , 接着, 两个{Si}5- 和溶液中的H+ 作用产生H2 [式(1~4)]。计算结果显示2 {Si }5- → {Si }6- + {Si }4- 需吸热15. 4kcal· mol- 1 , 使总能量达95. 4kcal· mol- 1 , 不是能量上有利的路径。然而, 质子化后, {Si(H)}4- 参与的反应(反应1 和8 )需要克服的总能垒为69. 1kcal· mol- 1 , 明显降低。实验上推测的OER中间体应为质子化中间体{Si(H)}4- 。接下来, 由{Si(H)}4- 生成的TER中间{Si(H2 ) }4- 裂解水释放氢气, 为有利的热力学放热过程(反应12 )。因此, 当满足多酸的垂直激发能时, 多酸裂解水是一个能量有利的过程。
李灿等[14 通过电化学实验发现{Si}4- 的四电子还原需要较高的外界能量, 因此, 在紫外-可见光区(λ>300nm), H2 来自于{Si}4- 的双电子还原。与Darwent等[12 的推测不同, 李灿等[14 推测TER中间体{Si}6- 来自于激发态的OER中间体{Si}5-* 。DFT计算结果显示该过程, 即反应2 和2′ 至少需要吸收能量62. 9kcal· mol- 1 。而且TDDFT计算的{Si }5- 的垂直激发能为39. 2kcal· mol- 1 , 说明即使有外界激发光的协助, 也不足以推动反应2 和2′ 发生。OER中间体应为来自耦合的电子和质子转移生成的{Si(H)}4- (反应1 )。接下来, 他们推测{Si}6- 和溶液中的H+ 作用生成H2 。由图 3 6- 至少需要克服95. 4kcal· mol- 1 的总能垒, 在能量上不是有利路径。计算结果显示质子化后的TER中间体{Si(H2 ) }4- 或{Si(H)}5- 的生成所需能量明显降低, 最低总能垒不超过63kcal· mol- 1 。因此, 实验上推测的TER中间体{Si(H2 ) }4- 或{Si(H)}5- 应来自于{Si}5- 或{Si(H)}4- 与甲醇自由基之间的电子和质子转移反应(反应4′ 和5′ ), 或来自于{Si(H)}4- 自身的电子和质子转移反应(反应8 )。最后, 由{Si(H2 ) }4- 或{Si(H)}5- 直接或裂解水释放H2 。计算结果显示这些反应均为无能垒的放热过程(反应11 和14 ), 这也符合Fortage等[18 的计算结果。
3.
结论
采用B3LYP杂化泛函, 对Si取代的Keggin型多酸[SiW12 O40 ]4- 催化光裂解水产氢机理进行了计算。结果显示整个光催化过程分为四个步骤:(1)光激发; (2)电荷转移和单电子还原OER中间体生成; (3)双电子TER中间体生成; (4) H2 释放和催化剂重生。多酸的光激发诱导电子从牺牲还原剂甲醇转移到多酸, 从而产生推动反应进行的活性推动力。一旦OER中间体生成, 后续TER中间体的生成和H2 产生均存在有利的热力学放热过程。可以看出, 多酸在整个光催化循环过程中, 与可见光相互作用发生光激发, 从牺牲还原剂或自身获得电子生成活性中间体(反应1~8′ ), 裂解水生成H2 (反应9~14 ), 扮演了光敏化剂、催化剂、电子的受体和给体。因耦合的电子和质子转移在整个催化过程中扮演了重要角色, 接下来将对多酸中心杂原子、多酸活性氧位点相连的金属原子、牺牲还原剂和溶剂的影响进行系统研究。

 下载:
下载:

 下载:
下载:






