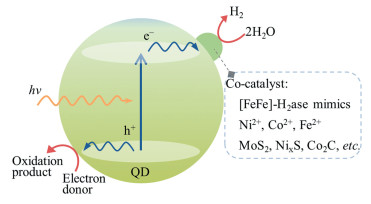
图 1.
量子点光催化制氢示意图
Figure 1.
Schematic illustration of photocatalytic hydrogen production from QDs

化石能源为人类社会发展做出巨大贡献,但是化石能源的不断开采和使用也带来诸多问题,如环境污染、全球气候变暖、能源危机等[1]。能源问题已经成为21世纪人类面临的首要课题,开发和利用可再生能源迫在眉睫[2]。太阳能作为可再生能源具有清洁、廉价、丰富等优点,自然界光合生物能够吸收太阳光还原质子生成H2或还原CO2生成有机物,将光能转化为化学能[3]。通过模拟自然界光合作用的机制,人工光合成也可以将太阳能转化为化学能[4]。例如,利用人工光合成分解水生成氢气和氧气,其中氢气作为清洁能源为人类提供燃料[5, 6]。
传统的光催化材料(如过渡金属配合物、块状半导体材料等)普遍存在易发生光腐蚀、合成复杂、禁带宽度大、活性低等问题[7],限制了这类光催化材料的实际应用。因此,进一步发展高效、稳定、制备方法简单的光催化材料意义重大。半导体量子点(Quantum dots,QDs)具有一系列特殊的光物理和光化学性质:(1)具有大的吸光系数和优异的可见光吸收能力;(2)由于量子点的尺寸小,光生电子和空穴向量子点表面的传输距离短,因而具有快速的电荷分离与传输性质;(3)多激子生成,量子点吸收高能光子后可能产生多个电子-空穴对,使光催化反应的量子效率大于100%[8];(4)可调的能带结构[9, 10],根据量子限域效应,可以通过调控量子点的尺寸改变其带隙宽度,拓宽量子点的吸光范围;(5)具有大的比表面积和丰富的表面活性位点;此外,(6)量子点合成方法简单,可以大规模水相制备。
半导体量子点已经成为人工光合成化学转化领域的明星材料,各类基于量子点的人工光合成体系被不断报道,在光催化分解水制氢、CO2还原、光催化有机转换等领域都展示出了优异的性能[11~14]。基于此,本文总结了笔者团队近期在量子点人工光合成领域的研究进展,并对该领域的发展进行了展望。
量子点人工光合成制氢过程包括光子吸收、光生载流子分离与传输、光生电子和光生空穴参与氧化还原催化反应三个步骤(图 1)。在众多组成不同的半导体量子点中,过渡金属-硫族化合物半导体量子点具有合适的带隙和导价带位置、优异的吸光能力,在光催化分解水制氢体系中得到广泛应用。此外,在光催化体系中合理引入制氢助催化剂,可以起到如下作用:(1)捕获光生电子,抑制光生电子和空穴的复合;(2)吸附与活化H2O分子,为光催化制氢反应提供活性位点;(3)降低质子还原反应活化能,加快催化反应动力学过程。
半导体量子点与仿生催化剂结合被证明是提高光催化制氢效率的有效方法。自2011年起,笔者团队以量子点(如CdTe QDs[15, 16]、CdSe QDs[17~21])为光敏剂、[FeFe]-氢化酶模拟物为助催化剂,构筑了一系列高效的光催化制氢体系,并将这类体系的产氢催化循环数(TON)提高到8.36×104[21]。进一步地,通过简单地向量子点体系中引入无机金属盐(如Co2+[22]、Ni2+[23~34]、Fe2+[35]等)可以原位构筑高效、稳定的人工光合成制氢催化剂,该方法已成为光解水制氢的一条高效、便捷且经济的途径。
然而,在真实催化反应条件下,催化前体的结构变化难以捉摸,人们对光催化析氢过程背后的机理知之甚少。针对这一难题,我们通过电子顺磁共振(EPR)和X射线吸收光谱(XAS)等原位表征手段,揭示了真实反应条件下活性位点的结构变化和光催化制氢动力学机制[34]。EPR和XAS测试表明,光照条件下,量子点受光激发生成的光生电子转移到NiⅡ,NiⅡ得到电子经NiI转变为Ni0团簇,从Ni(OH)2晶格中析出,形成具有丰富缺陷位点的[Nix0/Ni1-x(OH)2]。密度泛函理论(DFT)计算表明,原位生成的[Nix0/Ni1-x(OH)2]结构表面丰富的Ni空位能够有效地促进H2O分子的吸附和解离,提高局部质子浓度。与此同时,Ni0团簇作为真正的活性位点催化质子还原制氢。Ni0团簇与Ni空位协同作用,最终使体系在碱性水溶液中表现出高效的光催化制氢活性。
为了进一步提高光催化制氢效率,实现廉价金属离子助催化剂在量子点表面定点、定向、可控负载,加快光生电荷转移,我们在CdSe QDs表面生长了薄且各向异性的ZnS壳层,利用阳离子交换方法将部分Zn2+取代为M2+(M=Fe,Co,Ni等),成功制备得到集捕光单元、保护层、产氢助催化剂于一体的CdSe/Zn1-xMxS QDs[35]。ZnS壳层能够钝化CdSe表面缺陷位点,抑制光生激子在缺陷位点处的复合;同时作为中间介质将M2+助催化剂可控负载在极小的量子点表面。在可见光(λ=405nm)照下,CdSe/Zn1-xFexS QDs能够172h连续光催化产生H2(>880mL,39.3mmol),TON值高达6×105。
自然界高效进行的光合作用得益于生命体细胞内精密而复杂的组装体结构。受此启发,笔者团队模拟自然界光合作用反应中心,将多个量子点光催化剂与单个Pt助催化剂通过聚丙烯酸盐连接构筑人工光合组装体[36]。量子点与助催化剂间近距离的连接有利于多电子转移过程的发生,极大地提高了体系光催化制氢效率,基于Pt的TON值高达>1.64×107。在此基础上,我们通过可见光诱导需氧氧化的方法,在无外加添加剂的条件下,将Pt纳米颗粒与量子点组装形成微聚集体,促进量子点到助催化剂快速、持续的电子转移[37]。
二维层状材料具有大的比表面积和丰富的活性位点,有利于质子的吸附和还原,且超薄的平面结构具有短的载流子迁移距离和高的电荷迁移率,有利于光生激子的分离和传输。得益于此,我们利用CdSe/ZnS QDs和1T-MoS2纳米片之间的静电相互作用和配位作用构筑了QDs/1T-MoS2自组装结构[38]。基于该组装体的人工光合成制氢体系表现出优异的光催化制氢活性,在最优条件下氢气生成速率达~155±3.5μmol·h-1·mg-1。此外,以DFT计算为指导,合成了暴露特定晶面的Co2C纳米片,并通过自组装的方式与量子点结合建立了高效的光催化制氢体系[39]。
最近,笔者团队[40, 41]也逐步探索不外加制氢助催化剂,仅通过对量子点进行表面修饰实现了高效稳定的光催化产氢。例如,通过在CdSe QDs表面部分生长ZnS钝化量子点表面缺陷,能够显著提高量子点的光稳定性[40]。当ZnS的覆盖量为46%时光催化产氢TON值高达4.4×105,可与含产氢助催化剂的体系相媲美。
光(电)催化全分解水是一个热力学上坡反应,至少需要1.23eV的能量输入,且水氧化反应经历四电子转移过程[42]。在光催化分解水体系,过渡金属硫族化合物量子点容易被光生空穴氧化发生自身光腐蚀,利用量子点实现全解水极具挑战。Wolff等[43]在CdS纳米棒表面空间分离地负载产氢助催化剂和水氧化助催化剂,在可见光照射下同时放出氢气和氧气。受限于光生空穴对半胱氨酸配体和量子点晶格中S2-的氧化,未能实现化学计量比全分解水。光生空穴对过渡金属硫族化合物半导体量子点的氧化是这类体系实现化学计量比全分解水的主要瓶颈问题。李灿等[44]将光合系统Ⅱ(PSⅡ)和人工半导体光催化剂Ru2S3/CdS自组装构筑生物-人工杂化光催化全分解水体系,以Fe(CN)63-/Fe(CN)64-为电子中继体,实现了化学计量比全解水(411mol H2 ·(mol PSII)-1·h-1,205 mol O2·(mol PSII)-1·h-1)。当使用Ru/SrTiO3: Rh替换Ru2S3/CdS作为半导体光催化剂时,体系光催化全解水的活性显著提高(2489mol H2·(mol PSII)-1·h-1,1334mol O2·(mol PSII)-1·h-1),为进一步构建高效稳定的光催化全分解水体系提供了新的思路。
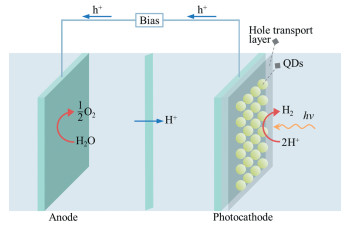
在量子点光电化学(PEC)分解水制氢中,量子点敏化光阴极受光激发生成的光生电子迁移至量子点表面活性位点还原质子产生氢气,光生空穴沿外电路迁移至对电极氧化水产生氧气(图 2)。光生电子与光生空穴的空间分离能够有效地抑制光生空穴对量子点的氧化腐蚀。PEC分解水制氢能够避免牺牲剂的使用,在阴极和阳极分别放出氢气和氧气。一般来说,量子点敏化光阴极体系的PEC分解水效率由吸光效率、光生激子分离和传输效率、氧化还原催化反应效率共同决定[45]。其中,由于量子点受光激发产生的光生空穴具有较大的有效质量,光生空穴传输速率远低于光生电子传输速率。光生空穴不能及时导出将促进光生载流子的非辐射跃迁,而且光生空穴对光生电子的库伦吸引作用将影响后续光生激子的分离。因此,实现光生空穴在电极界面快速传输是提高PEC分解水效率的关键。
2015年,笔者等[46]利用巯基乙酸(MAA)作为连接单元,实现CdSe QDs在NiO表面的组装,制备了量子点敏化光阴极。MAA配体不仅能够将量子点组装在NiO电极表面,而且能够作为空穴传输中继体促进界面间空穴传输,抑制光生载流子的复合。在可见光(λ>400nm)照射、-0.1V(vs. NHE)偏压、无外加助催化剂的条件下,CdSe QDs/NiO光阴极产生的光电流密度达-60μA·cm-2,且45h内光电流保持稳定,生成氢气的法拉第效率接近100%。多孔结构的NiO作为空穴传输层,不仅能增加量子点的负载量,提高体系对可见光的利用率,而且能够有效促进光生空穴的迁移,提高体系PEC分解水效率,使光阴极电流密度提高至-136μA·cm-2[47]。进一步地,通过在量子点表面引入空穴传输分子(吩噻嗪等),促进光生空穴在电极界面的传输,显著提升了光催化分解水制氢性能[48]。
2016年,笔者团队[49]利用石墨双炔(GDY)与量子点表面配体(4-巯基吡啶)之间强的π-π相互作用,将量子点负载于GDY表面,构筑量子点/GDY敏化光阴极。在0V(vs. NHE)电位下,CdSe QDs/GDY光阴极12h内光电流密度稳定在-70μA·cm-2,产氢法拉第效率为~95%。机理研究表明,GDY能使光生空穴快速地从量子点中导出,避免光生空穴在量子点处的积累和量子点的光腐蚀,从而提高了体系PEC分解水的活性和稳定性。
为了进一步增强体系对可见光的吸收、提高光生激子分离和界面间定向传输效率,通过量子点表面配体的配位作用连接CdSe QDs和CdTe QDs,形成量子点“手拉手”组装结构,并将其负载于NiO空穴传输层表面,构筑量子点敏化光阴极[50]。“手拉手”量子点组装结构不仅能够扩展体系的吸光范围,而且合适的能带结构使光生空穴在电极界面的传输能够直接、快速的进行。在-0.1V(vs. NHE)偏压下,“手拉手”CdSe-CdTe QDs敏化光阴极产生的光电流密度达到-140μA·cm-2,光电制氢法拉第效率为~92%。
尽管量子点敏化光阴极及PEC分解水已经取得了很大进展,但整体能量转化效率仍然较低,距大规模应用仍有一定的距离。从吸光、电荷分离及界面电荷传输、表面催化等方面,发展新的策略(如设计空穴传输层、表面钝化层、负载助催化剂、形成异质结等),建立无外加偏压、高效、稳定、廉价的PEC全分解水体系仍是未来努力的方向。
受制于水氧化极为不利的热力学和动力学因素,可见光驱动全解水制氢的能量转换效率普遍较低。因此,在光催化产氢的同时,克服水氧化的不利过程或避免使用牺牲剂,是构筑具有应用前景的人工光合成体系的关键。针对这一挑战,我们将人工光合成制氢或CO2还原与有机转换偶合起来,不仅能够实现太阳能到氢能的转化、闭合碳循环,而且能够利用空穴的氧化能力生产高附加值化学品(图 3)。
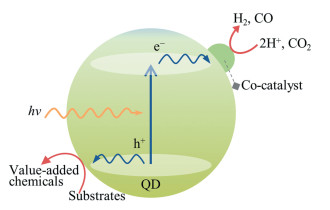
2014年,笔者等[51]利用CdSe QDs作为光催化剂,不外加氧化剂和电子牺牲体,实现了多种巯基化合物偶联生成二硫化合物,同时还原质子放出等当量的氢气。向体系中加入少量无机镍盐,可以显著提高二硫化合物和氢气生成速率和转化率。机理研究表明,由于巯基自由基与量子点结合力强,自偶联反应发生在量子点表面,而不是发生在溶液中;生成的二硫化合物与量子点结合力弱,容易从量子点表面脱附,可以避免二硫化合物的过氧化。进一步研究发现,CdSe QDs表面生成的巯基自由基还可以作为氧化剂选择性地氧化苄醇生成对应的醛/酮[52]。
与金属配合物、有机染料等均相光敏剂相比,将异相光催化剂半导体量子点用于光催化有机合成反应(如构筑S-S[51]、C-C[53~57]、C-N[56]键)具有无可比拟的优势(优异的吸光能力、可调的电子结构、出色的化学稳定性和丰富的活性位点等),近年来已经受到广泛关注[58~60]。Weix等[56]利用CdSe QDs实现了光催化β-烷基化、β-氨基烷基化、还原脱卤和脱羧自由基加成等反应,光催化效率媲美传统的均相光催化有机合成体系。Weiss等[53]利用CdSe QDs的三重态能量转移实现分子间可控的非对映选择性和区域选择性[2+2]环加成,为结构复杂的有机化合物的合成提供了新的思路。
最近,我们将CO2还原与有机转换相偶联,利用量子点受光激发生成的光生电子还原CO2生成CO,光生空穴氧化1-苯基乙醇生成对应的频哪醇[57]。在可见光(λ=450nm)照射下,CdSe/CdS QDs光催化CO生成速率为~27.63mmol·g-1·h-1,CO的选择性为~94.7%(竞争产物为H2),频哪醇的生成速率为~26.5mmol·g-1·h-1。13CO2同位素标记实验证明反应生成的CO仅来自气相中的CO2。机理研究指出,1-苯基乙醇首先吸附在量子点表面,被光生空穴氧化生成Cα自由基,Cα自由基在量子点表面偶联生成频哪醇,并从量子点表面解吸附。这是首次将可见光催化CO2还原与有机转换相偶联,不仅实现了CO2到CO的高效还原,同时在温和条件下合成了高附加值化学品。通过将人工光合成制氢/CO2还原与有机转换偶合,为建立更加高效、稳定、绿色、经济的人工光合成体系提供了新的思路。
量子点人工光合成化学转换是一项非常有前景的太阳能利用途径,它能够以充沛的H2O或CO2为原料,利用丰富的太阳能实现光(电)催化制氢、CO2还原、光催化有机转换等。半导体量子点光催化材料由于其独特的光物理性质,在人工光合成制氢/全解水/CO2还原领域已经取得一系列重要进展,但是仍面临许多挑战。建立更加高效、稳定的量子点人工光合成体系,进一步推动量子点在人工光合成领域的发展,需要以下几个方面的努力研究:(1)通过改变量子点的组成、形貌、尺寸等提高体系对可见光的利用率;(2)通过合理的引入助催化剂或空穴传输分子促进光生电子和空穴的分离和传输;(3)设计合适的活性位点提高量子点表面催化反应的活性和选择性;(4)实验与理论计算相结合,揭示催化反应动力学机制;(5)运用现代原位表征技术(如XPS、XAS、EPR等),获得真实反应条件下催化活性位点的结构变化,理解结构设计与光催化活性之间的关系;(6)通过深入且严谨的实验设计,结合ESR、原位漫反射傅里叶变换红外光谱(DRIFTS)、拉曼光谱、同位素标记实验、质谱、XAS等技术,获得光催化反应的中间体和反应路径,揭示光催化反应机理。我们相信,不远的将来在量子点人工光合成领域取得的巨大突破将为人类社会的可持续发展贡献重要力量。
Armaroli N, Balzani V. Angew. Chem. Int. Ed., 2007, 46(1/2): 52~66.
Gong J, Li C, Wasielewski M R. Chem. Soc. Rev., 2019, 48(7): 1862~1864. doi: 10.1039/C9CS90020A
Antal T K, Krendeleva T E, Rubin A B. Appl. Microbiol. Biotechnol., 2011, 89(1): 3~15. doi: 10.1007/s00253-010-2879-6
Whang D R, Apaydin D H. ChemPhotoChem, 2018, 2(3): 148~160. doi: 10.1002/cptc.201700163
Liu W, Cao L, Cheng W, et al. Angew. Chem. Int. Ed., 2017, 56(32): 9312~9317. doi: 10.1002/anie.201704358
Roger I, Shipman M A, Symes M D. Nat. Rev. Chem., 2017, 1(1): 0003. doi: 10.1038/s41570-016-0003
狄广兰, 朱志良. 化学通报, 2017, 80(3): 228~235. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20160927001&flag=1
Yan Y, Crisp R W, Gu J, et al. Nat. Energy, 2017, 2(5): 17052. doi: 10.1038/nenergy.2017.52
Robel I, Kuno M, Kamat P V. J. Am. Chem. Soc., 2007, 129(14): 4136~4137. doi: 10.1021/ja070099a
Kundu S, Patra A. Chem. Rev., 2017, 117(2): 712~757. doi: 10.1021/acs.chemrev.6b00036
Li X B, Tung C H, Wu L Z. Nat. Rev. Chem., 2018, 2(8): 160~173. doi: 10.1038/s41570-018-0024-8
陈雅静, 李旭兵, 佟振合, 等. 化学进展, 2019, 31(1): 38~49. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201901020.htm
Li X B, Tung C H, Wu L Z. Angew. Chem. Int. Ed., 2019, 58(32): 10804~10811. doi: 10.1002/anie.201901267
Wu H L, Li X B, Tung C H, et al. Adv. Mater., 2019, 31(36): 1900709. doi: 10.1002/adma.201900709
Wang F, Wang W G, Wang X J, et al. Angew. Chem. Int. Ed., 2011, 50(14): 3193~3197. doi: 10.1002/anie.201006352
Jian J X, Liu Q, Li Z J, et al. Nat. Commun., 2013, 4: 2695. doi: 10.1038/ncomms3695
Li C B, Li Z J, Yu S, et al. Energy Environ. Sci., 2013, 6(9): 2597~2602. doi: 10.1039/c3ee40992a
Wang F, Liang W J, Jian J X, et al. Angew. Chem. Int. Ed., 2013, 52(31): 8134~8138. doi: 10.1002/anie.201303110
Liang W J, Wang F, Wen M, et al. Chem. Eur. J., 2015, 21(8): 3187~3192. doi: 10.1002/chem.201406361
Jian J X, Ye C, Wang X Z, et al. Energy Environ. Sci., 2016, 9(6): 2083~2089. doi: 10.1039/C6EE00629A
Wen M, Li X B, Jian J X, et al. Sci. Rep., 2016, 6: 29851. doi: 10.1038/srep29851
Li Z J, Li X B, Wang J J, et al. Energy Environ. Sci., 2013, 6(2): 465~469. doi: 10.1039/C2EE23898E
Li Z J, Wang J J, Li X B, et al. Adv. Mater., 2013, 25(45): 6613~6618. doi: 10.1002/adma.201302908
Li Z J, Fan X B, Li X B, et al. J. Am. Chem. Soc., 2014, 136(23): 8261~8268. doi: 10.1021/ja5047236
Wang J J, Li Z J, Li X B, et al. ChemSusChem, 2014, 7(5): 1468~1475. doi: 10.1002/cssc.201400028
Fan X B, Yu S, Zhan F, et al. ChemSusChem, 2017, 10(24): 4833~4838. doi: 10.1002/cssc.201701950
Fan X B, Yu S, Wu H L, et al. J. Mater. Chem. A, 2018, 6(34): 16328~16332. doi: 10.1039/C8TA05637D
Huang M Y, Li X B, Gao Y J, et al. J. Mater. Chem. A, 2018, 6(14): 6015~6021. doi: 10.1039/C8TA00385H
Yu S, Fan X B, Wang X, et al. Nat. Commun., 2018, 9: 4009. doi: 10.1038/s41467-018-06294-y
Wang J J, Wang J, Feng K, et al. J. Mater. Chem. A, 2017, 5(20): 9537~9543. doi: 10.1039/C7TA00336F
Yu S, Li Z J, Fan X B, et al. ChemSusChem, 2015, 8(4): 642~649. doi: 10.1002/cssc.201402885
Li Z J, Fan X B, Li X B, et al. J. Mater. Chem. A, 2017, 5(21): 10365~10373. doi: 10.1039/C7TA01670K
Fan X B, Yu S, Wang X, et al. Adv. Mater., 2019, 31(7): 1804872. doi: 10.1002/adma.201804872
Wang Y, Ma Y, Li X B, et al. J. Am. Chem. Soc., 2020, 142(10): 4680~4689. doi: 10.1021/jacs.9b11768
Gao Y J, Li X B, Wang X Z, et al. Matter, 2020, 3(2): 571~585. doi: 10.1016/j.matt.2020.06.022
Li X B, Gao Y J, Wang Y, et al. J. Am. Chem. Soc., 2017, 139(13): 4789~4796. doi: 10.1021/jacs.6b12976
Wang Y, Li X B, Wu H L, et al. ACS Sustain. Chem. Eng., 2019, 7(7): 7286~7293. doi: 10.1021/acssuschemeng.9b00347
Li X B, Gao Y J, Wu H L, et al. Chem. Commun., 2017, 53(41): 5606~5609. doi: 10.1039/C7CC02366A
Guo Q, Liang F, Gao X Y, et al. ACS Catal., 2018, 8(7): 5890~5895. doi: 10.1021/acscatal.8b01105
Gao Y J, Li X B, Wu H L, et al. Adv. Funct. Mater., 2018, 28(33): 1801769. doi: 10.1002/adfm.201801769
Gao Y J, Yang Y C, Li X B, et al. Chem. Commun., 2018, 54(38): 4858~4861. doi: 10.1039/C8CC02091D
Bowker M. Green Chem., 2011, 13(9): 2235~2246. doi: 10.1039/c1gc00022e
Wolff C M, Frischmann P D, Schulze M, et al. Nat. Energy, 2018, 3(10): 862~869. doi: 10.1038/s41560-018-0229-6
Wang W, Chen J, Li C, et al. Nat. Commun., 2014, 5(1): 4647. doi: 10.1038/ncomms5647
Wu H L, Li X B, Tung C H, et al. Adv. Sci., 2018, 5(4): 1700684. doi: 10.1002/advs.201700684
Liu B, Li X B, Gao Y J, et al. Energy Environ. Sci., 2015, 8(5): 1443~1449. doi: 10.1039/C5EE00331H
Wen M, Wu H L, Li X B, et al. Part. Part. Syst. Charact., 2018, 35(1): 1700278~1700282. doi: 10.1002/ppsc.201700278
Li X B, Liu B, Wen M, et al. Adv. Sci., 2016, 3(4): 1500282. doi: 10.1002/advs.201500282
Li J, Gao X, Liu B, et al. J. Am. Chem. Soc., 2016, 138(12): 3954~3957. doi: 10.1021/jacs.5b12758
Wu H L, Li X B, Wang Y, et al. J. Mater. Chem. A, 2019, 7(45): 26098~26104. doi: 10.1039/C9TA10056C
Li X B, Li Z J, Gao Y J, et al. Angew. Chem. Int. Ed., 2014, 53(8): 2085~2089. doi: 10.1002/anie.201310249
Zhao L M, Meng Q Y, Fan X B, et al. Angew. Chem. Int. Ed., 2017, 56(11): 3020~3024. doi: 10.1002/anie.201700243
Jiang Y, Wang C, Rogers C R, et al. Nat. Chem., 2019, 11(11): 1034~1040. doi: 10.1038/s41557-019-0344-4
Zhang Z, Edme K, Lian S, et al. J. Am. Chem. Soc., 2017, 139(12): 4246~4249. doi: 10.1021/jacs.6b13220
Pal A, Ghosh I, Sapra S, et al. Chem. Mater., 2017, 29(12): 5225~5231. doi: 10.1021/acs.chemmater.7b01109
Caputo J A, Frenette L C, Zhao N, et al. J. Am. Chem. Soc., 2017, 139(12): 4250~4253. doi: 10.1021/jacs.6b13379
Guo Q, Liang F, Li X B, et al. Chem, 2019, 5(10): 2605~2616. doi: 10.1016/j.chempr.2019.06.019
Hao H, Lang X. ChemCatChem, 2019, 11(5): 1378~1393. doi: 10.1002/cctc.201801773
Lu H, Huang Z, Martinez M S, et al. Energy Environ. Sci., 2020, 13(5): 1347~1376. doi: 10.1039/C9EE03930A
Huang C, Li X B, Tung C H, et al. Chem. Eur. J., 2018, 24(45): 11530~11534. doi: 10.1002/chem.201800391

图 1 量子点光催化制氢示意图
Figure 1 Schematic illustration of photocatalytic hydrogen production from QDs

图 2 量子点敏化光阴极体系全分解水示意图
Figure 2 Schematic illustration of QDs photocathode system for overall photo-electrochemical water splitting

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
