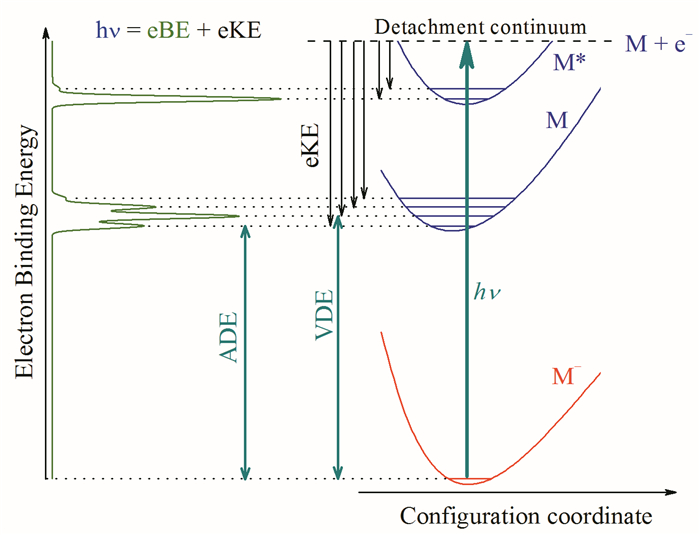
图 1.
负离子光电子能谱能量分析示意图
Figure 1.
Energetic principles of anion photoelectron spectroscopy

利用负离子光电子能谱(Anion photoelectron spectroscopy,aPES)技术可以探究诸多光谱和化学动力学方面的基础问题。几乎所有对应负离子能够产生且具有正的电子亲和势(Electron affinity,EA)的中性分子或团簇都能够利用aPES对其电子和几何结构进行研究。在aPES实验中,质量选择的单一质量的负离子与一束激光作用,如果激光光子能量超过电子结合为负离子的结合能,则会脱附出具有一定动能的光电子:
|
$ {{\rm{M}}^ - } + h\nu \to {\rm{M}} + {{\rm{e}}^ - } $ |
(1) |
其中,M-为我们研究的负离子,hν为激光的光子能量,M为产生的中性分子,e-为脱附光电子。可通过测量脱附光电子的动能分布揭示脱附到特定中性电子态和振动态的离散特征。光脱附过程的能量分析示意图如图 1所示。激光光子能量用以克服电子结合到给定中性态的结合能(eBE)和提供脱附后光电子的动能(eKE):
|
$ {\rm{eBE = }}h\nu - {\rm{eKE}} $ |
(2) |
光电子能谱通常以eBE为横坐标作图,因为其值与激光光子能量无关。通过aPES实验可以获得负离子及中性分子的许多信息。例如,可以获得中性分子基电子态及不同激发电子态的绝热电子脱附能(Adiabatic detachment energy,ADE)及垂直脱附能(Vertical detachment energy,VDE);当电子态存在振动分辨时,还可以获得相应电子态下分子的振动频率;某些情况下,还可能获得负离子的热带,从中可以获得负离子的振动频率。通常采用的脱附激光具有线偏振特征,在线性偏振光的单光子脱附过程中,电子速度成像方法可以同时获得表明光脱附过程电子来源轨道的对称性信息的光电子角分布信息[1]。
aPES可研究指定质量数的分子或团簇,从而可以探究团簇尺寸变化对电子和几何结构的影响[2~9],其中,Wang[10]对于特殊的硼烯及硼球烯结构的发现及其他硼团簇系列的探究促进了硼化学和纳米结构的研究。另外,可研究不同溶剂配体及不同溶剂分子数目对于负离子的微溶剂化效应[11~20]。M不必是稳定的,事实上也可以是双原子或单原子化学反应的过渡态,这种情况下,PES可以得到这些瞬态物种的振动能级,这在化学反应中至关重要[21~24]。
1967年,Brehm等[25]报道了第一次aPES实验,该实验利用连续离子源和连续波腔内氩离子激光器进行,并使用半球能量分析仪检测动能分布。随后,Lineberger等[26]运用该装置测量了多种主族元素及过渡金属的电子亲和势[27~32]。更短波长的脉冲激光器的发展以及脉冲式气流膨胀离子源的出现使得负离子的产生更为简便,研究者发展了脉冲负离子光电子能谱[33, 34]。起初,脱附电子动能分布的探测利用的是飞行时间(TOF)方法,脱附光可使用Nd:YAG激光器的多倍频[35, 36];最近有报道aPES使用了Nd:YAG激光器的九倍频,即波长为118nm(10.8eV)[37]。自由场式和磁瓶式TOF分析器都被广泛用来测量脱附电子动能分布。自由场式的分析器只能探测很小角度范围的光电子,其收集效率低,但有更高的分辨率,可达5~10 meV[33, 35]。磁瓶式TOF受多普勒展宽的影响,分辨率相对较低,为10~40 meV[38~41]。
一项重大的进步是Baguenard等[42]和Surber[43]将光电子速度成像(Velocity-map imaging,VMI)技术与aPES结合,这一结合的优势在于其有很高的收集效率且同时能得到光电子角分布信息。1987年,Chandler等[44, 45]首次运用离子速度成像方法研究CH3I光解离产物CH3碎片的二维离子影像,但其分辨能力较差。1997年,Eppink等[46]对成像透镜进行了重大改进,改进后的VMI技术极大提升了能量分辨率,ΔeKE/eKE约为3%。这样的分辨已足够测量部分振动分辨的电子跃迁,但是依旧不足以分辨低频振动和离子温度较高导致的密集光谱。
Muller-Dethlefs等[47, 48]将零动能能谱(Zero electron kinetic energy spectroscopy,ZEKE)技术应用在中性分子研究中,可以显著提高PES分辨率,后来Neumark等[49~51]将此技术用于负离子团簇的研究。在负离子零动能谱中,质量选择的负离子被一可调谐激光光脱附,随着激光波长扫描,只有那些在零动能附近的电子被收集。能量分辨可以达到1cm-1,但是对于多数分子,由于不能分辨的转动展宽,峰宽在8~10 cm-1[52]。
由于对于每一个激光波长只有很窄范围动能的电子被收集,所以采集一张全谱需要扫描步长很小的很多步;另外,零动能电子对杂散电场和磁场十分敏感,导致这种实验十分困难。另一方面,由于负离子的零动能和中性的零动能物理特性明显不同,负离子高里德堡态使得电子脱附不易进行[53],另外负离子零动能谱还受阈值定则的限制[54, 55],使得这项技术的应用受到很大限制。
上述传统的aPES采用固定脱附光波长,其探测效率非常高,零动能谱分辨率很高,但探测效率很低。Neumark等[56, 57]发展的慢电子速度成像技术(Slow electron velocity-map imaging,SEVI)结合了前面两种技术的优点,使得应用体系得到很大扩展。
该方法的基本过程是:利用波长可调谐的染料激光器,使脱附激光的光子能量略高于特定的跃迁阈值,这样脱附的电子动能范围仅有几十meV。这种技术只需采集不同波长下的光电子能谱,将其拼成一张完整的能谱图,便可以得到高分辨的光电子能谱,其收集效率接近100%。通常,由SEVI方法研究分子得到的光电子能谱的分辨率可达20~100 cm-1。
多电荷负离子在自然界普遍存在且是溶液相和固相的重要组成部分,在气相条件下,由于缺少溶剂分子的稳定作用,以及分子内多电荷间的库伦排斥,孤立的多电荷负离子变得十分脆弱。前期对于气相多电荷负离子的实验探究都是通过多种离子化方式利用质谱进行研究[58~63],但其光谱研究依旧是难题。尽管离子溅射、激光蒸发等碰撞技巧可以产生一些二价负离子,但其强度太弱不足以进行光谱研究。
由Yamashita等[64, 65]发展的电喷雾电离(ESI)技术作为一种软电离源,可直接从溶液中得到小的气相多电荷负离子,1998年Wang等[66~68]发展了一种结合了ESI和PES的实验技术来在气相中研究多电荷负离子。他们探究了多种多电荷负离子的性质,包括在PES中直接观察到多电荷负离子中普遍存在的库伦能垒[67, 68]。该技术对多电荷负离子的性质及其光电子能谱有深远的影响,它为多电荷负离子提供了动态稳定性。由于库伦能垒的存在,还可以形成一种有趣的亚稳态多电荷负离子,其电子结合能为负值,即其电子可以发生自发脱附,但由于库伦能垒的阻碍,亚稳态的多电荷负离子的寿命较长。对这类多电荷负离子进行光电子实验,得到的光电子的动能就会大于光子的能量[69, 70]。另外,研究者还利用此技术研究了多电荷负离子的溶剂化和溶剂稳定性[71~75]。
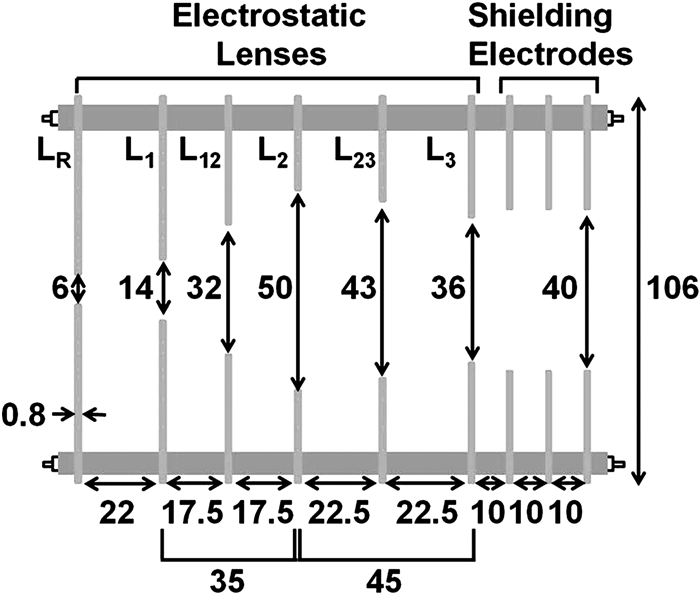
对于采用Eppink等设计的aPES,其分辨率约为3%,依旧有很大的提升空间。2007年,Cavanagh等[76]提出了一种具有很高分辨率的VMI透镜设计,对于0.87eV动能的电子,可达到ΔeKE/eKE=0.38%的能量分辨率,虽然未对其对于低能电子的分辨能力进行描述,但仍表明了该高分辨VMI透镜设计的潜力。2014年,在原本用于离子成像的四电极设计方案基础上[77],Wang等[78]报道了其新设计的VMI透镜,如图 2。该设计的优势在于其在保持对阈值附近电子高分辨能力(1.2cm-1)的情况下仍对高动能电子具有很高的分辨能力(ΔeKE/eKE=0.53%)。随后,Ning等[79~81]跟进了这一设计理念,并利用所搭建的高分辨慢电子速度成像仪精密测量了一系列原子的电子亲和势,将测量精度提高到大于0.1meV。Neumark等[82]随后也依据此改进了其SEVI透镜的设计。
限制PES分辨率的另一因素是常温下分子的振动、转动所导致的热展宽。2005年,Wang等[83]发展了具有温度控制能力的第二代电喷雾负离子光电子能谱仪,其关键点在于新增的3D Paul trap可以冷却和积累从ESI源产生的负离子,将负离子冷却至振动基态,减少分子的热展宽。低温离子阱技术的温度可调对于区分弱键合物种的异构体以及对复杂阴离子和生物分子的温度依赖性构象变化进行测定非常重要[84~86]。另外,Neumark[87]、Ning[88]、Von Issendorff[89]等课题组也已将离子阱冷却技术与原有光电子能谱仪结合,获得了优异的分辨能力,极大地扩展了光电子能谱技术的应用。
自aPES技术发展以来,诸多课题组利用此技术对于气相分子及团簇的电子结构及成键特性进行了大量研究[90~102],因篇幅原因,在此不再赘述。本文主要聚焦于aPES技术在研究锕系元素化合物及含铍化合物的电子特性中的应用。
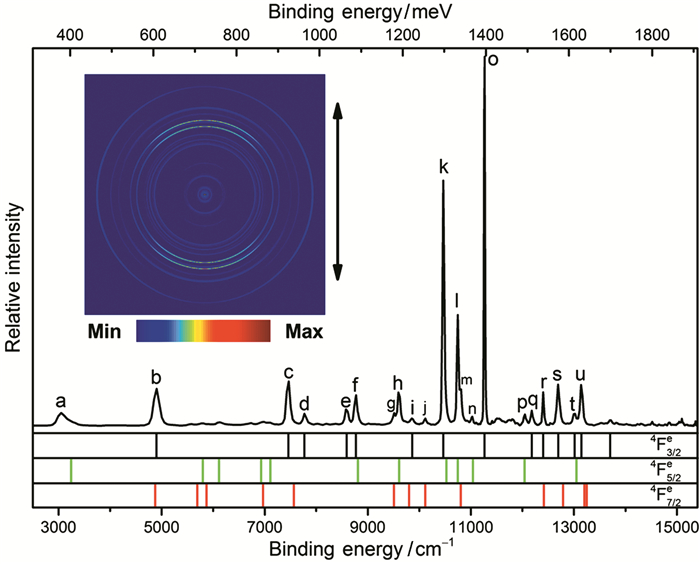
激光冷却是一种建立超冷中性原子或正离子集合的成熟技术[103~107]。在过去的40年中,这种技术开辟了许多令人兴奋的新研究领域。但是,到目前为止还没有带负电的离子被直接激光冷却。这是由于负离子独特的能级结构,其额外的电子和中性原子间的结合力很弱,因此负离子通常没有激发态,因而缺少激光冷却所需要的快速电偶极跃迁循环[108]。经过十多年的努力,La-成为最有前途的候选者[109]。笔者最近的研究表明,Th-是作为激光冷却的有希望的候选者[110]。Th-的高分辨光电子能谱见图 3。Th的实测和计算预测电子亲和力分别为4901.35(48)和4832 cm-1,或0.607690(60)和0.599 eV,几乎是以前的理论值0.368eV的两倍。Th-的基态确定为6d37s2 4Fe3/2,而不是6d27s27p4G°5/2。其结果是在Th-中,构型6d37s2和6d27s27p引起的束缚能级之间有几个强电偶极跃迁。潜在的激光冷却跃迁为2S°1/2
笔者课题组近期通过负离子光电子速度成像实验研究了ThO-、ThO2-和ThO3-,并结合Franck-Condon模拟和高精度量子化学计算进行了几何结构、电子结构、化学成键和谱峰归属分析[111, 112]。
实验测得的ThO分子的电子亲和能为0.707±0.020eV,通过Franck-Condon模拟得到ThO的振动频率为895(40)cm-1,ThO-的振动频率为810(40)cm-1。在充分考虑电子相关效应和标量及自旋-轨道耦合相对论效应的基础上,利用CASSCF/CASPT2/SO计算方法可精确求解ThO/ThO-的基态及激发态电子结构,进而准确解析实验谱图。计算结果与实验谱图的一致性表明负离子光电子速度成像实验方法结合量化计算是研究钍元素化合物的有效方法之一。在ThO-的光电子能谱中观察到了异常光电子角分布现象,在对脱附分子轨道的原子轨道组成进行计算后,将其视为“类原子轨道”,并采用Cooper-Zare方程对角分布进行了详细解释。此外,ThO-向ThO跃迁过程中存在着双电子跃迁现象,说明Th-7s和Th-6d电子间存在着强电子相关效应。
从实验中获得的ThO2的电子亲和能为1.21(5)eV,Franck-Condon模拟得到ThO2的振动频率是824(40)cm-1,ThO2-的振动频率是767(40)cm-1,结合理论计算分析了ThO2弯曲几何结构的原因;另外,从实验中观察到了ThO3-和ThO3间的跃迁通道,结合理论计算解析了ThO3和ThO3-的不同同分异构体。对于ThO3分子,仍需要进行高精度的实验探测以确定其电子亲和能。
通过对ThOx(x=1~3)体系的研究发现,随着O原子数目的增加,电子亲和能也在逐渐增加。化学键研究表明,Th主要以df杂化参与成键,在该体系中Th-O均为多重键,该计算结果和分析可帮助我们深入理解含钍化合物中Th-6d和Th-5f原子轨道间的杂化现象。
铍元素由于其广为人知的毒性,人们关于其化学性质知之甚少。铍元素最大的特点是作为碱土金属元素而具有独特的成键特性,目前,对于铍元素及含铍分子的相关实验研究开展较少。Heaven课题组[113~115]利用aPES技术研究了BeX-(X=O,S,F)双原子负离子的成键特性,结果表明,BeO和BeS相当离子化,EA值相对较高,而BeF的成键表现出明显的配位键特征,其EA值比BeO和BeS的一半还低;另外,因为BeO和BeS有足够大的偶极矩,可通过电荷-偶极作用束缚一个电子形成负离子的偶极束缚态(Dipole-bound state,DBS),但BeF因其较小的偶极矩,实验中未能观察到DBS的存在。
笔者课题组利用搭建的aPES仪对BeCl-负离子及其中性分子展开了研究,得到了其成像及能谱数据,如图 4。在光电子脱附的初态负离子及末态中性分子的结构变化很大,其很可能存在类似于BeF-负离子明显的配位键特征。另外,经过前期对不同靶材及配比、时序、溅射激光能量及气路压强的多次实验,已基本掌握了其他铍团簇及含铍化合物的产生条件,其质谱及光电子能谱探测实验将逐步展开。
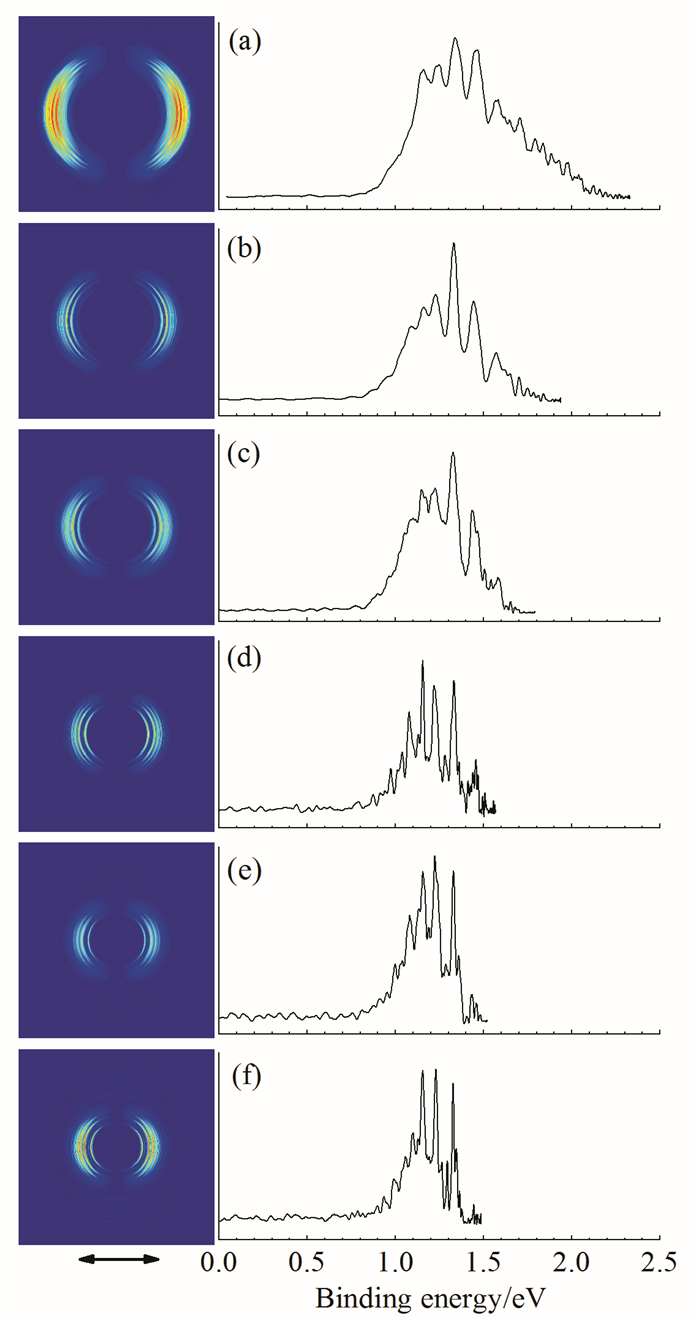
(脱附光能量为:(a)2.331eV;(b)1.937eV;(c)1.792eV;(d)1.567eV;(e)1.521eV;(f)1.485eV;双箭头表示脱附激光偏振方向)
本文简单综述了aPES技术自分辨率较低的传统飞行时间分析器到高分辨负离子速度成像技术的发展历程。简要分析了传统aPES、零动能谱及慢电子速度成像技术的优势与不足。电喷雾电离源与aPES的结合提供了一种通用且灵敏地在气相中探测多电荷负离子及溶液阴离子电子结构和稳定性的光谱学工具。高分辨速度成像透镜设计的提出和离子阱冷却的引入显著提高了光电子能谱的分辨能力。
笔者课题组探讨了aPES技术在锕系元素及含铍分子电子结构和化学键研究方面的应用。首次发现了负离子可作为激光冷却新的候选者,分析了ThOx(x=1~3)成键规律及BeX(X=O,S,F)的成键特征的异同。
Cooper J, Zare R N. J. Chem. Phys., 1968, 48(2):942~943. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Leopold D G, Ho J, Lineberger W C. J. Chem. Phys., 1987, 86(4):1715~1726. doi: 10.1063/1.452170
Li J, Li X, Zhai H J, et al. Science, 2003, 299(5608):864~867. doi: 10.1126/science.1079879
Häkkinen H, Yoon B, Landman U, et al. J. Phys. Chem. A, 2003, 107(32):6168~6175. doi: 10.1021/jp035437i
Shao N, Huang W, Gao Y, et al. J. Am. Chem. Soc., 2010, 132(18):6596~6605. doi: 10.1021/ja102145g
Pande S, Huang W, Shao N, et al. ACS Nano, 2016, 10(11):10013~10022. doi: 10.1021/acsnano.6b04330
Huang W, Ji M, Dong C D, et al. ACS Nano, 2008, 2(5):897~904. doi: 10.1021/nn800074b
Bulusu S, Li X, Wang L S, et al. PNAS, 2006, 103(22):8326~8330. doi: 10.1073/pnas.0600637103
Kostko O, Huber B, Moseler M, et al. Phys. Rev. Lett., 2007, 98(4):043401. doi: 10.1103/PhysRevLett.98.043401
Wang L S. Int. Rev. Phys. Chem., 2016, 35(1):69~142. doi: 10.1080/0144235X.2016.1147816
Markovich G, Pollack S, Giniger R, et al. J. Chem. Phys., 1994, 101(11):9344~9353. doi: 10.1063/1.467965
Wang X B, Yang X, Wang L S, et al. J. Chem. Phys., 2002, 116(2):561~570. doi: 10.1063/1.1427067
Wang X B, Yang X, Nicholas J B, et al. J. Chem. Phys., 2003, 119(7):3631~3640. doi: 10.1063/1.1590641
Hendricks J H, de Clercq H L, Freidhoff C B, et al. J. Chem. Phys., 2002, 116(18):7926~7938. doi: 10.1063/1.1457444
Arnold S T, Hendricks J H, Bowen K H. J. Chem. Phys., 1995, 102(1):39~47. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Mabbs R, Surber E, Sanov A. J. Chem. Phys., 2005, 122(5):54308. doi: 10.1063/1.1839861
Sobhy M A, Casalenuovo K, Reveles J U, et al. J. Phys. Chem. A, 2010, 114(42):11353~11363. doi: 10.1021/jp1058148
Castleman A, Bowen K. J. Phys. Chem., 1996, 100(31):12911~12944. doi: 10.1021/jp961030k
Schiedt J, Weinkauf R, Neumark D M, et al. Chem. Phys., 1998, 239(1-3):511~524. doi: 10.1016/S0301-0104(98)00361-9
Verlet J, Bragg A, Kammrath A, et al. Science, 2005, 307(5706):93~96. doi: 10.1126/science.1106719
Polanyi J C, Zewail A H. Acc. Chem. Res., 2002, 28(3):119~132. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Yacovitch T I, Garand E, Kim J B, et al. Faraday Discus., 2012, 157:399~414. doi: 10.1039/c2fd20011b
Gómez H, Meloni G, Madrid J, et al. J. Chem. Phys., 2003, 119(2):872~879. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Yang X, Wang X B, Wang L S. J. Chem. Phys., 2001, 115(7):2889~2892. doi: 10.1063/1.1394758
Brehm B, Gusinow M A, Hall J L. Phys. Rev. Lett., 1967, 19(13):737~741. doi: 10.1103/PhysRevLett.19.737
Hotop H, Bennett R A, Lineberger W C. J. Chem. Phys., 1973, 58(6):2373~2378. doi: 10.1063/1.1679514
Feigerle C S, Corderman R R, Lineberger W C. J. Chem. Phys., 1981, 74(2):1513~1515. doi: 10.1063/1.441174
Feigerle C S, Corderman R R, Bobashev S V, et al. J. Chem. Phys., 1981, 74(3):1580~1598. doi: 10.1063/1.441289
Leopold D G, Lineberger W C. J. Chem. Phys., 1986, 85(1):51~55. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Kasdan A, Lineberger W C. Phys. Rev. A, 1974, 10(5):1658~1664. doi: 10.1103/PhysRevA.10.1658
Kasdan A, Herbst E, Lineberger W C. J. Chem. Phys., 1975, 62(2):541~548. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Miller T M, Miller A E, Lineberger W C. Phys. Rev. A, 1986, 33(5):3558~3559. doi: 10.1103/PhysRevA.33.3558
Posey L A, Deluca M J, Johnson M A. Chem.Phys. Lett., 1986, 131(3):170~174. doi: 10.1016/0009-2614(86)80539-5
Cheshnovsky O, Yang S, Pettiette C, et al. Chem. Phys. Lett., 1987, 138(2-3):119~124. doi: 10.1016/0009-2614(87)80353-6
Waller I, Kitsopoulos T, Neumark D. J. Phys. Chem., 1990, 94(6):2240~2242. doi: 10.1021/j100369a009
Cheshnovsky O, Taylor K J, Conceicao J, et al. Phys. Rev. Lett., 1990, 64(15):1785~1788. doi: 10.1103/PhysRevLett.64.1785
Yang J, Wang X B, Xing X P, et al. J. Chem. Phys., 2008, 128(20):201102. doi: 10.1063/1.2938390
Wang L S, Cheng H S, Fan J. J. Chem. Phys., 1995, 102(24):9480~9493. doi: 10.1063/1.468817
Handschuh H, Ganteför G, Eberhardt W. Rev. Sci. Instrum., 1995, 66(7):3838~3843. doi: 10.1063/1.1145446
Giniger R, Hippler T, Ronen S, et al. Rev. Sci. Instrum., 2001, 72(6):2543~2549. doi: 10.1063/1.1367364
Thomas O C, Zheng W, Bowen K H. J. Chem. Phys., 2001, 114(13):5514~5519. doi: 10.1063/1.1349547
Baguenard B, Pinare J, Lepine F, et al. Chem. Phys. Lett., 2002, 352(3-4):147~153. doi: 10.1016/S0009-2614(01)01449-X
Surber E, Sanov A. J. Chem. Phys., 2002, 116(14):5921~5924. doi: 10.1063/1.1467916
Chandler D W, Houston P L. J. Chem. Phys., 1987, 87(2):1445~1447. doi: 10.1063/1.453276
Helm H, Bjerre N, Dyer M J, et al. Phys. Rev. Lett., 1993, 70(21):3221~3224. doi: 10.1103/PhysRevLett.70.3221
Eppink A T J B, Parker D H. Rev. Sci. Instrum., 1997, 68(9):3477~3484. doi: 10.1063/1.1148310
Müller-Dethlefs K, Sander M, Schlag E W. Chem. Phys. Lett., 1984, 112(4):291~294. doi: 10.1016/0009-2614(84)85743-7
Muller-Dethlefs K, Schlag E W. Ann. Rev. Phys. Chem., 1991, 42(1):109~136. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Arnold C C, Neumark D M, Cyr D M, et al. J. Phys. Chem., 1995, 99(6):1633~1636. doi: 10.1021/j100006a002
Kitsopoulos T, Waller I, Loeser J, et al. Chem. Phys. Lett., 1989, 159(4):300~306. doi: 10.1016/0009-2614(89)87488-3
Metz R, Weaver A, Bradforth S, et al. J. Phys. Chem., 1990, 94(4):1377~1388. doi: 10.1021/j100367a034
Lenzer T, Yourshaw I, Furlanetto M R, et al. J. Chem. Phys., 1999, 110(19):9578~9586. doi: 10.1063/1.478923
Simons J. J. Phys. Chem. A, 2008, 112(29):6401~6511. doi: 10.1021/jp711490b
Reed K J, Zimmerman A H, Andersen H C, et al. J. Chem. Phys., 1976, 64(4):1368~1375. doi: 10.1063/1.432404
Wigner E P. Phys. Rev., 1948, 73(9):1002~1009. doi: 10.1103/PhysRev.73.1002
Osterwalder A, Nee M J, Zhou J, et al. J. Chem. Phys., 2004, 121(13):6317~6322. doi: 10.1063/1.1787491
Neumark D M. J. Phys. Chem. A, 2008, 112(51):13287~13301. doi: 10.1021/jp807182q
Schauer S N, Williams P, Compton R N. Phys. Rev. Lett., 1990, 65(5):625~628. doi: 10.1103/PhysRevLett.65.625
Middleton R, Klein J. Nucl. Instrum. Meth. B, 1997, 123(1-4):532~538. doi: 10.1016/S0168-583X(96)00734-3
Calabrese D, Covington A M, Thompson J S. J. Chem. Phys., 1996, 105(7):2936~2937. doi: 10.1063/1.472156
Klein J, Middleton R. Nucl. Instrum. Meth. B, 1999, 159(1-2):8~21. doi: 10.1016/S0168-583X(99)00178-0
Middleton R, Klein J. Phys. Rev. A, 1999, 60(5):3515. doi: 10.1103/PhysRevA.60.3515
Gnaser H. Phys. Rev. A, 1999, 60(4):R2645. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Yamashita M, Fenn J B. J. Phys. Chem., 1984, 88(20):4451~4459. doi: 10.1021/j150664a002
M Yamashita, J B Fenn. J. Phys. Chem., 1984, 88(20):4671~4675. doi: 10.1021/j150664a046
Wang L S, Ding C F, Wang X B, et al. Rev. Sci. Instrum., 1999, 70(4):1957~1966. doi: 10.1063/1.1149694
Wang X B, Ding C F, Wang L S. Phys. Rev. Lett., 1998, 81(16):3351. doi: 10.1103/PhysRevLett.81.3351
Wang L S, Ding C F, Wang X B, et al. Phys. Rev. Lett., 1998, 81(13):2667. doi: 10.1103/PhysRevLett.81.2667
Wang X B, Wang L S. Nature, 1999, 400(6741):245~248. doi: 10.1038/22286
Wang X B, Wang L S. Phys. Rev. Lett., 1999, 83(17):3402. doi: 10.1103/PhysRevLett.83.3402
Wang X B, Nicholas J B, Wang L S. J. Chem. Phys., 2000, 113(24):10837~10840. doi: 10.1063/1.1333703
Wang X B, Yang X, Nicholas J B, et al. Science, 2001, 294(5545):1322~1325. doi: 10.1126/science.1064916
Wang X B, Yang X, Wang L S. Int. Rev. Phys. Chem., 2010, 21(3):473~498. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Yang X, Fu Y J, Wang X B, et al. J. Am. Chem. Soc., 2004, 126(3):876~883. doi: 10.1021/ja038108c
Minofar B, Mucha M, Jungwirth P, et al. J. Am. Chem. Soc., 2004, 126(37):11691~11698. doi: 10.1021/ja047493i
Cavanagh S J, Gibson S T, Gale M N, et al. Phys. Rev. A, 2007, 76(5):052708. doi: 10.1103/PhysRevA.76.052708
Townsend D, Minitti M P, Suits A G. Rev. Sci. Instrum., 2003, 74(4):2530~2539. doi: 10.1063/1.1544053
Leon I, Yang Z, Liu H T, et al. Rev. Sci. Instrum., 2014, 85(8):083106. doi: 10.1063/1.4891701
Chen X, Ning C. Phys. Rev. A, 2016, 93(5). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Chen X, Luo Z, Li J, et al. Sci. Rep., 2016, 6:24996. doi: 10.1038/srep24996
Chen X, Ning C. J. Chem. Phys., 2016, 145(8):084303. doi: 10.1063/1.4961654
Weichman M L, DeVine J A, Levine D S, et al. PNAS, 2016, 113(7):1698~1705. doi: 10.1073/pnas.1520862113
Wang X B, Wang L S. Rev. Sci. Instrum., 2008, 79(7):073108. doi: 10.1063/1.2957610
Wang X B, Woo H K, Wang L S. J. Chem. Phys., 2005, 123(5):051106. doi: 10.1063/1.1998787
Woo H K, Wang X B,Wang L S, et al. J. Phys. Chem. A, 2005, 109(47):10633~10637. doi: 10.1021/jp0553277
Woo H K, Wang X B, Kiran B, et al. J. Phys. Chem. A, 2005, 109(50):11395~11400. doi: 10.1021/jp0529467
Hock C, Kim J B, Weichman M L, et al. J. Chem. Phys., 2012, 137(24):244201. doi: 10.1063/1.4772406
Luo Z, Chen X, Li J, et al. Phys. Rev. A, 2016, 93(2):020501. doi: 10.1103/PhysRevA.93.020501
Bartels C, Hock C, Kuhnen R, et al. J. Phys. Chem. A, 2014, 118(37):8270~8276. doi: 10.1021/jp5010902
Wang L S. Phys. Chem. Chem. Phys., 2010, 12(31):8694~8705. doi: 10.1039/c003886e
Gao Y, Huang W, Woodford J, et al. J. Am. Chem. Soc., 2009, 131(27):9484~9485. doi: 10.1021/ja903043d
Cui L F, Wang L S. Int. Rev. Phys. Chem., 2008, 27(1):139~166. doi: 10.1080/01442350701791256
Pande S, Jian T, Khetrapal N S, et al. J. Phys. Chem. C, 2018, 122(12):6947~6954. doi: 10.1021/acs.jpcc.8b00166
Li W L, Chen X, Jian T, et al. Nat. Rev. Chem., 2017, 1(10):0071. doi: 10.1038/s41570-017-0071
Weichman M L, Neumark D M. Ann. Rev. Phys. Chem., 2018, 69:101~124. doi: 10.1146/annurev-physchem-050317-020808
Young R M, Neumark D M. Chem. Rev., 2012, 112(11):5553~5577. doi: 10.1021/cr300042h
Liu G, Zhu Z, Ciborowski S M, et al. Angew. Chem., 2019, 58(23):7773~7777. doi: 10.1002/anie.201903252
Visser B R, Addicoat M A, Gascooke J R, et al. J. Chem. Phys., 2016, 145(4):044320. doi: 10.1063/1.4959130
Hirata K, Tomihara R, Kim K, et al. Phys. Chem. Chem. Phys., 2019, 21(32):17463~17474. doi: 10.1039/C9CP02622C
Wu X, Tan K, Tang Z, et al. Phys. Chem. Chem. Phys., 2014, 16(10):4771~4777. doi: 10.1039/c3cp51851e
Liu J-X, Liu Z, FilotI A W, et al. Catal, Sci, Technol,, 2017, 7(1):75~83. doi: 10.1039/C6CY02277D
Felton J, Ray M, Jarrold C C. Phys, Rev, A, 2014, 89(3):033407. doi: 10.1103/PhysRevA.89.033407
Ashkin A. Phys. Rev. Lett., 1978, 40(12):729~732. doi: 10.1103/PhysRevLett.40.729
Wineland D J, Drullinger R E, Walls F L. Phys. Rev. Lett., 1978, 40(25):1639~1642. doi: 10.1103/PhysRevLett.40.1639
Neuhauser W, Hohenstatt M, Toschek P, et al. Phys. Rev. Lett., 1978, 41(4):233~236. doi: 10.1103/PhysRevLett.41.233
Wieman C E, Pritchard D E, Wineland D J. Rev. Modern Phys., 1999, 71(2):S253. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Cronin A D, Schmiedmayer J, Pritchard D E. Rev. Modern Phys., 2009, 81(3):1051~1129. doi: 10.1103/RevModPhys.81.1051
Andersen T. Phys. Rep., 2004, 394(4-5):157~313. doi: 10.1016/j.physrep.2004.01.001
Cerchiari G, Kellerbauer A, Safronova M S, et al. Phys. Rev. Lett.,2018, 120(13):133205. doi: 10.1103/PhysRevLett.120.133205
Tang R, Si R, Fei Z, et al. Phys. Rev. Lett., 2019, 123:203002. doi: 10.1103/PhysRevLett.123.203002
Li Y, Zou J, Xiong X G, et al. J. Phys. Chem. A, 2017, 121(10):2108~2113. doi: 10.1021/acs.jpca.6b11554
Li Y, Zou J, Xiong X G, et al. J. Chem. Phys., 2018, 148(24):244304. doi: 10.1063/1.5030142
Mascaritolo K J, Dermer A R, Green M L, et al. J. Chem. Phys., 2017, 146(5):054301. doi: 10.1063/1.4974843
Dermer A R, Green M L, Mascaritolo K J, et al. J. Phys. Chem. A, 2017, 121(30):5645~5650. doi: 10.1021/acs.jpca.7b04894
Green M L, Jean P, Heaven M C. J. Phys. Chem. Lett., 2018, 9(8):1999~2002. doi: 10.1021/acs.jpclett.8b00784



图 4 不同脱附光能量下得到的BeCl-的光电子成像及能谱图
Figure 4 Photoelectron images and spectra of BeCl- obtained at (a)2.331eV, (b) 1.937eV, (c) 1.792eV, (d) 1.567eV, (e) 1.521eV, (f) 1.485eV. The double arrow indicates the direction of laser polarization
(脱附光能量为:(a)2.331eV;(b)1.937eV;(c)1.792eV;(d)1.567eV;(e)1.521eV;(f)1.485eV;双箭头表示脱附激光偏振方向)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载: