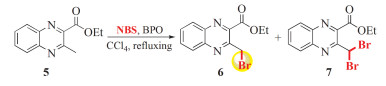
Scheme 1.
化合物5的自由基溴化反应
Scheme 1.
The radical bromination reaction of 5 with NBS
Convenient and Effective Syntheses of Novel 3-Aroxymethylquinoxaline-2-carboxylic acids and Their Antibacterial Activities
Hong Zhang , Yunhe He , Yang Li , Yu Chen

It has been well established that quinoxaline ring system is an exceptional class of nitrogen-containing heterocycles, which present in many pharmacologically relevant molecules with significant antibacterial, antimicrobial, antimalarial and anti-cancer properties, etc[1~4]. In this regard, Ajani highlighted the status of quinoxaline motifs as excellent pathfinders in therapeutic medicine[5]. Recently, Tariq et al.[6] have reviewed the advance on pharmacological activities of quinoxaline derivatives. Due to the diverse striking biological properties, syntheses of suitably substituted quinoxalines for drug design have received much attention in the field of contemporary medicinal chemistry[7~9]. Especially, in the past decade, the syntheses and antibacterial activities of 2-substituted and 2, 3-disubstituted quinoxaline derivatives have been frequently reported by both organic and medicinal chemists with the aim of developing new and effective antibacterial agents[10, 11]. For example, Xia et al.[12] have described recently the synthesis and potent antibacterial activity of quinoxaline-based chalcone derivatives (1).
|
|
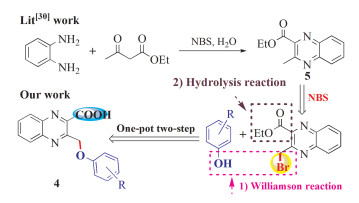
On the other hand, Williamson reaction, which usually involves the reaction between alkyl halide and alkali-metal salt of the hydroxy compounds,[13] is a very useful transformation in organic synthesis since the products widely exist as substructures in numerous medicinally interesting compounds and are of value in both industrial and academic applications[14, 15]. For example, 2-(aroxymethyl) quinolines like PF2545920 (2)[16] and RG5901 (3)[17] derived from the Williamson reaction of 2- halomethylquinoline with phenols displayed potent biological and pharmacological activities. As a consequence, much synthetic efforts have been devoted surrounding the Williamson reaction for the development of bioactive molecules by both organic and medicinal chemists[18~20]. Additionally, Maresca et al.[21] have found that many carboxylic acids incorporating various aromatic/heterocyclic scaffold showed significant potential for developing antibacterial agents with a diverse mechanism of action compared to the clinically used drugs.
Pertaining to these findings, we felt it would be an attractive molecular template by combination of the structural features of quinoxaline, aroxymethyl moiety and carboxylic acid functional group in a single framework, because it might lead to a new dimension of structural diversity as potential candidates for current medicinal chemistry needs. Accordingly, in consonance with our continued interest in the syntheses of structurally novel and intriguing Nheterocyclic compounds[22~29], we would like to report herein the simple and facile syntheses and antibacterial activities of a series of structurally new 3-aroxymethylquinoxaline-2-carboxylic acids (4) using ethyl 3-bromomethyl quinoxaline-2-carboxylate as key substrate via Williamson ether synthesis followed by ester hydrolysis reaction sequence.
The chemicals used in this work were obtained from Energy Chemical and were used without purification. Melting points were determined by use of a WRS-1B melting point apparatus without temperature correction. The1H (400MHz) and 13C (101MHz) NMR spectra were recorded on an Agilent 400-MR spectrometer using DMSO-d6 as the solvent. The reported chemical shifts (δ values) were given in parts per million downfield from tetramethylsilane (TMS) as the internal standard. HRMS (ESI) data were acquired on a Bruker Customer micrOTOF-Q 125 highresolution mass spectrometer with ESI. Elemental analyses were carried out on an EA 2400II elemental analyzer (PerkinElmer, Waltham, MA). The progress of reactions was monitored by TLC on silica gel GF254 using ethyl acetate/petroleum ether (1 : 8) as the eluent.
Ethyl 3-methylquinoxaline-2-carboxylate (5) (10. 812 g, 50 mmol) was added to CCl4(500 mL) and refluxed gently with stirring. A catalytic amount of benzoyl peroxide (BPO) (0. 302 g, 1. 25 mmol) as initiator was then added to the reaction mixture. After that, a slightly excessive amount of NBS (10. 678 g, 60 mmol) was added carefully in three batches to the gently refluxing CCl4 solution, i. e., 3. 56 g (20 mmol) portions of 10. 678 g (60 mmol) NBS were added every 1. 5 ~ 2. 0 h. After complete addition, the mixture continued to reflux gently till the disappearance of 1 (as monitored by TLC). The reaction mixture was cooled to room temperature and the precipitated succinimide was filtered off. The obtained filtrate was washed with water and dried over Na2 SO4. Evaporation of the solvent under reduced pressure afforded a crude solid product, which was subjected to column chromatography over silica gel (200 ~ 400 mesh) using petroleum ether/ethyl acetate mixture (10 :1, v/v) as eluent to give 10. 46 g of product 6 and 3. 37 g of 7.
Ethyl 3-(bromomethyl) quinoxaline-2- carboxylate (6) : white solid; m. p. 96 ~ 98℃; yield 71%;1 H NMR (400MHz, CDCl3) δ: 1. 54 (t, J = 7. 2 Hz, 3H, OCH2 CH3), 4. 63 (q, J = 7. 2 Hz, 2H, OCH2CH3), 5. 16 (s, 2H, CH2Br), 7. 86 ~ 7. 91 (m, 2H, Quin-H), 8. 12 (d, J = 7. 2 Hz, 1H, Quin-H), 8. 25 (d, J = 7. 2 Hz, 1H, Quin-H).13C NMR (101MHz, CDCl3) δ: 14. 20, 31. 54, 62. 98, 128. 99, 129. 88, 131. 27, 132. 41, 140. 59, 141. 87, 143. 62, 151. 23, 164. 85. HRMS: Calcd. For: C12H11 79BrN2 NaO2 [M + Na]+ : 316. 9897, Found: 316. 9906. Anal Calcd for C12H11BrN2 O2 : C, 48. 84; H, 3. 76; N, 9. 49%. Found: C, 48. 56; H, 3. 87; N, 9. 78%.
A mixture of ethyl 3-(bromomethyl) quinoxaline-2-carboxylate (6) (0. 295 g, 1. 0 mmol), respective substituted phenol 8a ~ 8l (1. 2 mmol), and anhydrous K2CO3 (0. 552 g, 4. 0 mmol) was stirred in CH3CN (15 ml) under reflux for 3 h. The conversion was monitored by TLC. When the reaction was complete, CH3CN was removed and a solution of KOH (1. 96 g, 35. 0 mmol) in 80% ethanol (20 mL) was directly added to the residue. The resulting reaction mixture was continued to heat under reflux for 2 h. After completion, the reaction mixture was cooled and acidified with 1 mol/L hydrochloric acid solution. The resulting crude product was recrystallized from ethanol to afford the pure products. The yields, physical properties and the spectral and analytical data are given below.
3-(Phenoxymethyl) quinoxaline-2-carboxylic acid (4a). White solid, m. p. 117 ~ 119℃, yield 87%;1 H NMR (400MHz, DMSO-d6) δ: 5. 92 (s, 2H, CH2), 6. 85 (t, J = 7. 6 Hz, 1H, Ben-H), 6. 97 (d, J = 8. 4 Hz, 1H, Ben-H), 7. 08 ~ 7. 14 (m, 2H, Ben-H), 7. 78 ~ 7. 92 (m, 3H, Ben-H and Quin-H), 8. 07 (d, J = 7. 6 Hz, 1H, QuinH), 8. 14 (d, J = 8. 4 Hz, 1H, Quin-H), 14. 29 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 69. 71, 112. 27, 120. 75, 126. 69, 129. 03, 129. 43, 130. 61, 130. 65, 131. 60, 139. 56, 143. 13, 151. 97, 157. 03, 164. 94. HRMS: Calcd. For: C16H12N2NaO3 [M + Na]+ : 303. 0741, Found: 303. 0726. Anal. Calcd for C16 H12 N2 O3 : C, 68. 56; H, 4. 32; N, 9. 99%; Found: C, 68. 74; H, 4. 16; N, 9. 91%.
3-((o-Tolyloxy) methyl) quinoxaline-2- carboxylic acid (4b). White solid, m. p. 125 ~ 126℃, yield 84%;1H NMR (400MHz, DMSO-d6) δ: 2. 16 (s, 3H, Me), 5. 58 (s, 2H, CH2), 6. 87 (t, J = 7. 6 Hz, 1H, Ben-H), 7. 07 (d, J = 8. 4 Hz, 1H, Ben-H), 7. 14 ~ 7. 17 (m, 2H, Ben-H), 7. 96 ~ 8. 02 (m, 2H, Quin-H), 8. 18 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 8. 22 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 14. 03 (s br, 1H, COOH); 13C NMR (101MHz, DMSO-d6) δ: 26. 52, 65. 26, 107. 21, 116. 59, 121. 86, 122. 76, 124. 63, 125. 03, 126. 36, 127. 23, 127. 98, 135. 78, 136. 67, 141. 86, 146. 17, 151. 98, 162. 48. HRMS: Calcd. For: C17H14N2NaO3 [M + Na]+ : 317. 0897, Found: 317. 0870. Anal. Calcd for C17 H14N2O3: C, 69. 38; H, 4. 79; N, 9. 52%; Found: C, 69. 59; H, 4. 86; N, 9. 38%.
3-((p-Tolyloxy) methyl) quinoxaline-2- carboxylic acid (4c). White solid, m. p. 123 ~ 124℃, yield 91%;1H NMR (400MHz, DMSO-d6) δ: 2. 23 (s, 3H, Me), 5. 54 (s, 2H, CH2), 6. 93 (d, J = 8. 4 Hz, 2H, Ben-H), 7. 10 (d, J = 8. 0 Hz, 2H, Ben-H), 7. 95 ~ 8. 01 (m, 2H, QuinH), 8. 16 (dd, J = 7. 6, 2. 4 Hz, 1H, Quin-H), 8. 21 (dd, J = 7. 6, 2. 4 Hz, 1H, Quin-H), 14. 01 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 15. 93, 65. 24, 110. 50, 124. 61, 125. 04, 125. 67, 125. 76, 127. 16, 127. 95, 135. 68, 136. 70, 141. 68, 146. 25, 151. 87, 162. 40. HRMS: Calcd. For: C17 H14N2NaO3 [M + Na]+ : 317. 0897, Found: 317. 0902. Anal. Calcd for C17H14N2O3: C, 69. 38; H, 4. 79; N, 9. 52%; Found: C, 69. 19; H, 4. 68; N, 9. 67%.
3-((3-Methoxyphenoxy) methyl) quinoxaline-2-carboxylic acid (4d). White solid, m. p. 149 ~ 151℃, yield 86%;1 H NMR (400MHz, DMSO-d6) δ: 3. 75 (s, 3H, OMe), 5. 59 (s, 2H, CH2), 6. 58 (d, J = 8. 4 Hz, 1H, Ben-H), 6. 62 (s, 1H, Ben-H), 6. 64 (d, J = 8. 8 Hz, 1H, Ben-H), 7. 22 (t, J = 8. 0 Hz, 1H, BenH), 7. 97 ~ 8. 03 (m, 2H, Quin-H), 8. 19 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 8. 23 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 14. 02 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 50. 93, 65. 24, 96. 95, 102. 69, 112. 75, 124. 61, 125. 05, 125. 86, 127. 21, 128. 00, 135. 71, 136. 72, 141. 61, 146. 06, 155. 19, 156. 26, 162. 37. HRMS: Calcd. For: C17 H14N2NaO4 [M + Na]+ : 333. 0846, Found: 333. 0870. Anal. Calcd for C17H14N2O4: C, 65. 80; H, 4. 55; N, 9. 03%; Found: C, 66. 04; H, 4. 29; N, 9. 31%.
3-((2-(tert-Butyl) phenoxy) methyl) quinoxaline-2-carboxylic acid (4e). White solid, m. p. 180 ~ 181℃, yield 81%;1 H NMR (400MHz, DMSO-d6) δ: 1. 30 (s, 9H, t-Bu), 5. 66 (s, 2H, CH2), 6. 91 (t, J = 7. 6 Hz, 1H, Ben-H), 7. 13 (d, J = 8. 0 Hz, 1H, Ben-H), 7. 20 (t, J = 8. 0 Hz, 1H, Ben-H), 7. 25 (d, J = 7. 6 Hz, 1H, BenH), 7. 97 ~ 8. 03 (m, 2H, Quin-H), 8. 17 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 8. 25 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 14. 03 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 25. 53, 30. 25, 65. 22, 108. 46, 116. 53, 122. 18, 123. 01, 124. 65, 125. 16, 127. 23, 128. 19, 133. 20, 135. 73, 137. 04, 140. 67, 146. 48, 152. 97, 162. 08. HRMS: Calcd. For: C20H20N2NaO3 [M + Na]+ : 359. 1367, Found: 359. 1381. Anal. Calcd for C20 H20N2O3: C, 71. 41; H, 5. 99; N, 8. 33%; Found: C, 71. 62; H, 6. 13; N, 8. 18%.
3-((4-(tert-Butyl) phenoxy) methyl) quinoxaline-2-carboxylic acid (4f). White solid, m. p. 184 ~ 185℃, yield 83%;1 H NMR (400MHz, DMSO-d6) δ: 1. 27 (s, 9H, t-Bu), 5. 56 (s, 2H, CH2), 6. 97 (d, J = 8. 8 Hz, 2H, Ben-H), 7. 33 (d, J = 8. 8 Hz, 2H, Ben-H), 7. 98 ~ 8. 04 (m, 2H, Quin-H), 8. 19 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 8. 23 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 14. 04 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 27. 15, 29. 66, 65. 20, 110. 04, 121. 99, 124. 63, 125. 04, 127. 19, 127. 96, 135. 71, 136. 69, 139. 21, 141. 79, 146. 24, 151. 72, 162. 41. HRMS: Calcd. For: C20 H20N2NaO3 [M + Na]+ : 359. 1367, Found: 359. 1376. Anal. Calcd for C20H20N2O3: C, 71. 41; H, 5. 99; N, 8. 33%; Found: C, 71. 13; H, 6. 10; N, 8. 62%.
3-((2-Fluorophenoxy) methyl) quinoxaline- 2-carboxylic acid (4g). Yellow solid, m. p. 169 ~ 170℃, yield 72%;1H NMR (400MHz, DMSO-d6) δ: 5. 70 (s, 2H, CH2), 6. 95 ~ 7. 00 (m, 1H, Ben-H), 7. 11 (t, J = 7. 6 Hz, 1H, Ben-H), 7. 22 ~ 7. 30 (m, 2H, Ben-H), 7. 95 - 8. 01 (m, 2H, Quin-H), 8. 14 (dd, J = 8. 0, 2. 0 Hz, 1H, QuinH), 8. 22 (dd, J = 8. 0, 2. 0 Hz, 1H, Quin-H), 14. 01 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 65. 96, 111. 36, 111. 92 (J = 17. 7 Hz), 117. 47 (J = 6. 8 Hz), 120. 56 (J = 3. 7 Hz), 124. 82 (J = 49. 4 Hz), 127. 24, 128. 10, 135. 75, 136. 77, 140. 98, 141. 94 (J = 10. 4 Hz), 145. 87, 146. 38, 148. 81, 162. 20. HRMS: Calcd. For: C16H11FN2NaO3 [M + Na]+ : 321. 0646, Found: 321. 0649. Anal. Calcd for C16H11FN2 O3 : C, 64. 43; H, 3. 72; N, 9. 39%; Found: C, 64. 21; H, 3. 87; N, 9. 57%.
3-((2-Chlorophenoxy) methyl) quinoxaline- 2-carboxylic acid (4h). White solid, m. p. 133 ~ 135℃, yield 75%;1H NMR (400MHz, DMSO-d6) δ: 5. 57 (s, 2H, CH2), 6. 86 (td, J = 7. 6, 1. 2 Hz, 1H, Ben-H), 6. 94 (td, J = 7. 6, 1. 2 Hz, 1H, Ben-H), 7. 00 (dd, J = 7. 6, 1. 2 Hz, 1H, Ben-H), 7. 08 (dd, J = 7. 6, 1. 2 Hz, 1H, BenH), 7. 96 ~ 8. 02 (m, 2H, Quin-H), 8. 16 (dd, J = 8. 0, 2. 4 Hz, 1H, Quin-H), 8. 22 (dd, J = 8. 0, 2. 4 Hz, 1H, Quin-H), 14. 04 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 66. 17, 108. 31, 110. 44, 116. 48, 117. 75, 124. 62, 125. 08, 127. 21, 128. 04, 135. 73, 136. 80, 141. 43, 143. 52, 145. 25, 146. 35, 162. 28. HRMS: Calcd. For: C16H1135ClN2 NaO3 [M + Na]+ : 337. 0351, Found: 337. 0347. Anal. Calcd for C16H11ClN2 O3 : C, 61. 06; H, 3. 52; N, 8. 90%; Found: C, 61. 17; H, 3. 43; N, 9. 07%.
3-((4-Bromophenoxy) methyl) quinoxaline- 2-carboxylic acid (4i). Yellow solid, m. p. 158 ~ 159℃, yield 87%;1H NMR (400MHz, DMSO-d6) δ: 5. 52 (s, 2H, CH2), 6. 92 (d, J = 8. 4 Hz, 2H, Ben-H), 7. 38 (d, J = 8. 8 Hz, 2H, Ben-H), 7. 86 ~ 7. 91 (m, 2H, Quin-H), 8. 05 (d, J = 8. 4 Hz, 1H, Quin-H), 8. 12 (d, J = 8. 4 Hz, 1H, Quin-H), 13. 96 (s br, 1H, COOH);13 C NMR (101MHz, DMSO-d6) δ: 69. 93, 113. 04, 117. 54, 129. 19, 129. 68, 131. 80, 132. 59, 132. 65, 140. 28, 141. 30, 145. 80, 150. 52, 157. 92, 166. 90. HRMS: Calcd. For: C16H1179BrN2 NaO3 [M + Na]+ : 380. 9846, Found: 380. 9857. Anal. Calcd for C16H11BrN2 O3 : C, 53. 50; H, 3. 09; N, 7. 80%; Found: C, 53. 73; H, 3. 34; N, 7. 62%.
3-((4-(tert-Butyl) -2-fluorophenoxy) methyl) quinoxaline-2-carboxylic acid (4j). White solid, m. p. 189 ~ 190℃, yield 81%;1 H NMR (400MHz, DMSO-d6) δ: 1. 26 (s, 9H, t-Bu), 5. 66 (s, 2H, CH2), 7. 11 (dd, J = 8. 4, 1. 2 Hz, 1H, Ben-H), 7. 19 (t, J = 8. 8 Hz, 1H, Ben-H), 7. 26 (dd, J = 13. 6, 1. 2 Hz, 1H, BenH), 7. 98 ~ 8. 04 (m, 2H, Quin-H), 8. 19 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 8. 24 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 14. 05 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 26. 89, 29. 87, 66. 12, 109. 18 (J = 18. 1 Hz), 110. 81, 116. 74 (J = 3. 1 Hz), 124. 64, 125. 11, 127. 27, 128. 10, 135. 77, 136. 77, 139. 45 (J = 10. 8 Hz), 140. 71 (J = 5. 1 Hz), 141. 18, 145. 97 (J = 3. 2 Hz), 148. 41, 162. 22. HRMS: Calcd. For: C20H19FN2NaO3 [M + Na]+ : 377. 1272, Found: 377. 1287. Anal. Calcd for C20H19 FN2 O3 : C, 67. 79; H, 5. 40; N, 7. 91%; Found: C, 67. 96; H, 5. 26; N, 8. 14%.
3-((4-(tert-Butyl) -2-chlorophenoxy) methyl) quinoxaline-2- carboxylic acid (4k). White solid, m. p. 192 ~ 193℃, yield 74%;1 H NMR (400MHz, DMSO-d6) δ: 1. 26 (s, 9H, t-Bu), 5. 68 (s, 2H, CH2), 7. 20 (d, J = 8. 8 Hz, 1H, Ben-H), 7. 30 (dd, J = 8. 8, 2. 0 Hz, 1H, Ben-H), 7. 42 (d, J = 2. 0 Hz, 1H, Ben-H), 7. 98 ~ 8. 04 (m, 2H, Quin-H), 8. 18 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 8. 24 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 13. 99 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 26. 89, 29. 81, 66. 08, 109. 67, 116. 87, 120. 69, 122. 72, 124. 64, 125. 12, 127. 29, 128. 13, 135. 81, 136. 76, 140. 56, 141. 16, 145. 92, 147. 20, 162. 15. HRMS: Calcd. For: C20H1935 ClN2NaO3 [M + Na]+ : 393. 0977, Found: 393. 0956. Anal. Calcd for C20H19ClN2 O3 : C, 64. 78; H, 5. 16; N, 7. 55%; Found: C, 65. 01; H, 5. 41; N, 7. 40%.
3-((2-Bromo-4-(tert-butyl) phenoxy) methyl) quinoxaline-2-carboxylic acid (4l). White solid, m. p. 195 ~ 196℃, yield 78%;1 H NMR (400MHz, DMSO-d6) δ: 1. 27 (s, 9H, t-Bu), 5. 63 (s, 2H, CH2), 7. 05 (d, J = 8. 4 Hz, 1H, Ben-H), 7. 19 (dd, J = 8. 4, 2. 0 Hz, 1H, Ben-H), 7. 27 (d, J = 2. 0 Hz, 1H, Ben-H), 7. 98 ~ 8. 04 (m, 2H, Quin-H), 8. 18 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 8. 25 (dd, J = 7. 6, 2. 0 Hz, 1H, Quin-H), 13. 96 (s br, 1H, COOH);13C NMR (101MHz, DMSO-d6) δ: 29. 51, 33. 63, 69. 21, 111. 93, 122. 79, 123. 20, 128. 52, 129. 00, 131. 05, 131. 96, 136. 28, 139. 63, 140. 89, 142. 13, 144. 84, 150. 44, 154. 61, 165. 98. HRMS: Calcd. For: C20H1979 BrN2NaO3 [M + Na]+ : 437. 0472, Found: 437. 0448. Anal. Calcd for C20H19BrN2 O3 : C, 57. 84; H, 4. 61; N, 6. 75%. Found: C, 57. 73; H, 4. 79; N, 7. 03%.
All the newly-synthesized compounds (4a ~ 4l) herein were screened for their potential in vitro antibacterial activities against Bacillus subtilis (B. subtilis) [CMCC (B) 63501], Staphylococcus aureus (S. aureus) [CMCC (B) 26003], Escherichia coli (E. coli) [CMCC (B) 44102] and Pseudomonas aeruginosa (P. aeruginosa) [CMCC (B) 10104] and anti-tubercular activity against Mycobacterium smegmatis (M. smegmatis) [CGMCC 1. 2621] by the broth microdilution assay. Each of the test compounds was dissolved in DMSO and then was serially diluted in five concentrations at 2-fold dilutions (250, 125, 62. 5, 31. 25, 15. 625, 7. 8125, 3. 90625 μg/mL) to determine the MICs. Rifampicin and Ciprofloxacin were used as the reference standards.
In the previous work, Kumar et al.[30]reported a facile and convenient synthesis of ethyl 3- methylquinoxaline-2-carboxylate (5) through the NBS-mediated reaction of ethyl acetoacetate with phenylene diamine (Scheme 1). To our knowledge, however, the further transformation by NBS radical bromination reaction at its 3-methyl position has never been reported. It is well known that halomethyl-functionalized aromatic heterocycles have been widely served as versatile building blocks for the construction of various intriguing and complex small molecules[31~33]. Thus, in this perspective, we envisioned that if the functionalization of 5 at its 3-methyl position with bromomethyl moiety could be achieved, the resulting ethyl 3-(bromomethyl) quinoxaline-2-carboxylate might open opportunity for the construction of the targeted compounds 4 through the radiacal bromination reaction followed by the one-pot sequential Williamson reaction/hydrolysis reaction procedure as depicted in Scheme 1.
Accordingly, with this assumption in mind, the first stage to implement our strategy was the radical bromination reaction of 5. Thus, the substrate 5, readily available following the protocol of the literature,[30] was subjected to the standard radical bromination reaction conditions together with 1. 2 equiv. of NBS in refluxing CCl4 with the presence of catalytic amount of benzoyl peroxide (BPO) as initiator as shown in Scheme 2.
We found that the addition order of NBS had an obvious influence on the product yield. When the amount of 1. 2 equiv. of NBS was added simultaneously with the substrate 5 to CCl4 solution, the radical bromination reaction suffered from low yield because the desired monobromo product 6 was always accompanied by the excess gem-dibromo byproduct 7 and small amount of unreacted substrate 5. Moreover, due to the close polarities of the three compounds, the isolation of them became very tedious and cumbersome. This is a problem that is common to numerous radical bromination protocols[34]. In order to characterize the structures of 6 and 7, we attempted to isolate them by careful column chromatography over silica gel using petroleum ether/ethyl acetate mixture as eluent (15 :1, v/v). After repeating the column chromatography separation three times, the desired monobrominated product 6 was obtained in 49% yield, along with 30% of the dibromide byproduct 7, 12% unreacted 5 as well as small quantities of tarry products.
We found that if the NBS was added in batches in the amount of 1/3 portions of the 1. 2 equiv. every 1. 5 ~ 2. 0h to the gently refluxing CCl4 solution, a 22% increase in the product yield to 71% was achieved with a small amount of byproduct after the reaction was complete. Presumably, the resulting Br2 derived from NBS remain in a low concentration throughout the course of the bromination reaction, thereby restraining the side reaction and leading to the formation of the desired product in a higher yield. The newly resulting 6 represents a valuable scaffold and will be highly useful in the field of synthetic organic chemistry. Its structure was easily confirmed by the1H and13C NMR spectral data. In its1H NMR spectrum, no signal attributable to the methyl protons of its precursor was observed, but instead a two-proton singlet at δ 5. 16 was found, readily recognizable as arising from bromomethyl protons, supporting the signal of its13C NMR spectrum at δ 62. 98 for the methylene carbon. Similarly, the structure of the dibromide byproduct 7 was also readily established according to its spectral data (see Experimental section).
Having the newly-synthesized ethyl 3- bromomethyl-quinoxaline-2-carboxylate (6) in hand, our attention was turned to its Williamson reaction with various phenols available in our lab for building the desired 3-aroxymethylquinoxaline-2- carboxylic acids (4). Accordingly, our investigation toward its Williamson reaction with phenols was conducted using MeCN as the solvent with K2CO3 as the base as shown in Scheme 3. Gratifyingly, the Williamson reaction proceeded very smoothly, and the TLC analysis did not indicate the formation of any distinct by-product after the substrates were completely consumed within 3h.
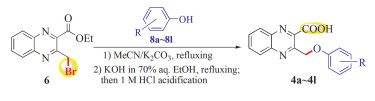
At this stage, we conceived that the Williamson reaction conditions might not interfere with subsequent ester hydrolysis reaction. Thus, upon completion of the Williamson reaction, the solvent MeCN was evaporated to dryness under reduced pressure, and the in situ ester hydrolysis reaction was conducted by refluxing the residue in 70% aq. ethanolic KOH solution for 2 h. After the alkaline hydrolysis reaction was complete followed by acidifying with 1 mol/L HCl, the desired 3- aroxymethylquinoxaline-2-carboxylic acids (4a ~ 4l) were obtained in overall good yields of 72% ~ 91% after recrystallization from ethanol as listed in Table 1. To the best of our knowledge, all these newly synthesized compounds 4a ~ 4l have never been reported, and their structures were explicitly characterized based on the spectral and analytical data. As an example, the1H NMR spectrum of 4a was devoid of the singlet signal of bromomethyl protons and the triplet and quartet signal set due to the ethyl ester protons, but instead contained a readily recognizable methylene proton singlet at δ 5. 92 and a broad carboxyl proton singlet at δ 14. 29, along with the signals for 9 aromatic protons between δ 6. 85 and 8. 14, which is consistent with the introduction of the nascent phenoxymethyl moiety to the quinoxaline ring.
 下载:
导出CSV
下载:
导出CSV
| Entry | Compd. | Yield/%a | B. subtilis | S. aureus | E. coli | P. aeruginosa | M. smegmatis |
| 1 |  |
87 | 125 | 125 | 125 | 250 | 250 |
| 2 |  |
84 | 250 | 250 | 250 | 250 | 250 |
| 3 |  |
91 | 250 | 250 | 250 | 250 | 250 |
| 4 |  |
86 | 250 | 125 | 125 | 125 | 250 |
| 5 |  |
81 | 125 | 125 | 250 | 125 | 250 |
| 6 |  |
83 | 62.5 | 62.5 | 250 | 250 | 250 |
| 7 |  |
72 | 62.5 | 31.25 | 125 | 125 | 250 |
| 8 |  |
75 | 125 | 62.5 | 125 | 125 | 250 |
| 9 |  |
87 | 62.5 | 62.5 | 125 | 250 | 125 |
| 10 |  |
81 | 15.625 | 7.8125 | 125 | 125 | 250 |
| 11 |  |
74 | 62.5 | 31.25 | 125 | 250 | 250 |
| 12 |  |
78 | 31.25 | 15.625 | 125 | 250 | 125 |
| 13 | Ciprofoxacin | - | 15.625 | 15.625 | 15.625 | 15.625 | - |
| 14 | Rifampicin | - | - | - | - | - | 15.625 |
| a Isolated yields | |||||||
As we all know, due to the serious concern related to the resistance of pathogenic bacteria towards the clinically used antibacterial drugs, screening new class of compounds for development of new antibacterial drugs is an urgent priority to overcome the increasing danger of drug-resistant problems. Investigations aimed at developing new antibacterial reagents have been carried out in our laboratory[29], and we have especially focused on the synthesis of structurally diverse small molecules. Therefore, after successfully synthesizing a series of the desired quinoxalines 4a ~ 4l, a preliminary evaluation for their in vitro antibacterial activities against Bacillus subtilis (B. subtilis) and Staphylococcus aureus (S. aureus) as Gram (+), Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa) as Gram (-) and Mycobacterium smegmatis (M. smegmatis) was assayed by measuring minimum inhibitory concentrations (MICs), and the test results were also recorded in Tab. 1.
From Tab. 1 it was observed that the compounds 4j, 4k and 4l with tert-butyl and halo (F, Cl, Br) substitution showed significant antibacterial activities against the tested Gram (+) bacterials S. aureus and B. subtilis (Entries 10 ~ 12), among them 4j had the best activity with the MIC values of 15. 625 and 7. 8125 μg/mL, respectively (Entry 10), better than the MIC value of the reference drug ciprofoxacin (15. 625 μg/mL). The insight would provide valuable information for further optimization of the series of derivatives, and hopefully contribute to the development of new and effective antibacterial agent. Our next efforts will mainly focus on the structural activity relationship study by structural optimization and exploring more structural diversity towards the ultimate goal of providing intriguing lead compounds for the development of new and effective antibacterial agents.
In conclusion, we have achieved a facile synthesis of a new series of structurally intriguing 3- aroxymethylquinoxaline-2-carboxylic acids (4a ~ 4l) through the radical bromination reaction followed by the one-pot sequential Williamson reaction/hydrolysis reaction procedure. This method features simple experimental operations, inexpensive reagents and high yields. The target compounds were evaluated for in vitro antibacterial activities against five bacterial strains, and the results revealed that compounds with both tert-butyl and halo (F, Cl and Br) substituents were promising with respect to the inhibitory activity against Gram (+) bacterials B. subtilis and S. aureus, among these compounds 4j possessed the best activity with the MIC values of 15. 625 and 7. 8125 μg/mL, respectively, being equipotent or even better than the reference drug Ciprofoxacin. Further research is currently ongoing in our laboratory, mainly focusing on the structural optimization and application of this method for the synthesis of other bioactive compounds, which will be communicated in due course.
Kaushal T, Srivastava G, Sharma A, et al. Bioorg. Med. Chem., 2019, 27(1): 16~35. doi: 10.1016/j.bmc.2018.11.021
Jaso A, Zarranz B, Aldana I, et al. J. Med. Chem., 2005, 48(6): 2019~2025. doi: 10.1021/jm049952w
El-Atawy M A, Hamed E A, Alhadi M, et al. Molecules, 2019, 24: 4198~4214. doi: 10.3390/molecules24224198
El-Attar M A Z, Elbayaa R Y, Shaaban O G, et al. Bioorg. Chem., 2018, 76: 437~448. doi: 10.1016/j.bioorg.2017.12.017
Ajani O O. Eur. J. Med. Chem., 2014, 85: 688~715. doi: 10.1016/j.ejmech.2014.08.034
Tariq S, Somakala K, Amir M. Eur. J. Med. Chem., 2018, 143: 542~557. doi: 10.1016/j.ejmech.2017.11.064
Chan C K, Chang M Y. Synthesis, 2016, 48(21): 3785~3793. doi: 10.1055/s-0035-1561472
Azab I H El, Elkanzi N A, Gobouri A A. J. Heterocycl. Chem., 2018, 55(1): 65~76. doi: 10.1002/jhet.2978
Lima R N, Porto A L. Tetrahed. Lett., 2017, 58(9): 825~828. doi: 10.1016/j.tetlet.2016.12.062
Kumar K S, Rambabu D, Sandra S, et al. Bioorg. Med. Chem., 2012, 20(5): 1711~1722. doi: 10.1016/j.bmc.2012.01.012
Cogo J, Kaplum V, Sangi D P, et al. Eur. J. Med. Chem., 2015, 90: 107~123. doi: 10.1016/j.ejmech.2014.11.018
Xia R, Guo T, He J, et al. Monatsh. Chem., 2019, 150: 1325~1334. doi: 10.1007/s00706-019-02449-9
Fuhrmann E, Talbiersky J. Org. Proc. Res. Dev., 2005, 9(2): 206~211. doi: 10.1021/op050001h
Tang L, Tian M, Chen H, et al. Dyes Pigments, 2018, 158: 482~489. doi: 10.1016/j.dyepig.2017.12.028
Guillén-Castellanos S A, Parent J S, Whitney R A. J. Polym. Sci. A, 2006, 44(2): 983~992. doi: 10.1002/pola.21221
Verhoest P R, Chapin D S, Corman M, et al. J. Med. Chem., 2009, 52(16): 5188~5196. doi: 10.1021/jm900521k
Galemmo Jr R A, Johnson Jr W H, Learn K S, et al. J. Med. Chem., 1990, 33(10): 2828~2841. doi: 10.1021/jm00172a024
Shibatomi K, Kotozaki M, Sasaki N, et al. Chem. Eur. J., 2015, 21(40): 14095~14098. doi: 10.1002/chem.201502042
Matsuya Y, Suzuki N, Kobayashi S, et al. Bioorg. Med. Chem., 2010, 18: 1477~1481. doi: 10.1016/j.bmc.2010.01.014
Li Z, Yang J, Gu W, et al. RSC Adv., 2016, 6(52): 46356~46365. doi: 10.1039/C6RA07356E
Maresca A, Vullo D, Scozzafava A, et al. J. Enzym. Inhib. Med. Chem., 2013, 28(2): 392~396. doi: 10.3109/14756366.2011.650168
Gao W, Fu X, Zhang X, et al. Tetrahed. Lett., 2016, 57: 4145~4148. doi: 10.1016/j.tetlet.2016.07.109
Li Y, Li K, Gao W. Chem. Heterocycl. Compd., 2016, 52(3): 200~205. doi: 10.1007/s10593-016-1856-0
Li Y, Wang Y, Zou H. Mol. Divers., 2017, 21(2): 463~473. doi: 10.1007/s11030-017-9730-2
Li Y, Zou H. J. Heterocycl. Chem., 2018, 55(1): 346~350. doi: 10.1002/jhet.3052
Li Y. Res. Chem. Intermed., 2015, 41(7): 4977~4985. doi: 10.1007/s11164-014-1581-1
高文涛, 符鑫博, 李阳.化学通报, 2016, 79(7): 630~639. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20151027003&flag=1
王东方, 符鑫博, 赵雅楠, 等.化学通报, 2017, 80(1): 69~76. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20160520002&flag=1
Li Y, Xu Q, Li Z, et al. Mol. Divers., 2020, 24(1): 167~178. doi: 10.1007/s11030-019-09938-3
Kumar B S P A, Madhav B, Reddy K H V, et al. Tetrahed. Lett. 2011, 52(22): 2862~2865.
Ghate M, Manohar D, Kulkarni V, et al. Eur. J. Med. Chem., 2003, 38: 297~302. doi: 10.1016/S0223-5234(03)00016-3
Young G L, Smith S A, Taylor R J. Tetrahed. Lett., 2004, 45(19): 3797~3801. doi: 10.1016/j.tetlet.2004.03.083
Aljaar N, Conrad J, Beifuss U. J. Org. Chem., 2013, 78(3): 1045~1053. doi: 10.1021/jo302491x
Thapa R, Brown J, Balestri T, et al. Tetrahed. Lett., 2014, 55(49): 6743~6746. doi: 10.1016/j.tetlet.2014.08.069
Table 1. Yields and in vitro antibacterial activity of the compounds 4a ~ 4l [MIC/(μg· mL-1)]
| Entry | Compd. | Yield/%a | B. subtilis | S. aureus | E. coli | P. aeruginosa | M. smegmatis |
| 1 | |
87 | 125 | 125 | 125 | 250 | 250 |
| 2 | |
84 | 250 | 250 | 250 | 250 | 250 |
| 3 | |
91 | 250 | 250 | 250 | 250 | 250 |
| 4 | |
86 | 250 | 125 | 125 | 125 | 250 |
| 5 | |
81 | 125 | 125 | 250 | 125 | 250 |
| 6 | |
83 | 62.5 | 62.5 | 250 | 250 | 250 |
| 7 | |
72 | 62.5 | 31.25 | 125 | 125 | 250 |
| 8 | |
75 | 125 | 62.5 | 125 | 125 | 250 |
| 9 | |
87 | 62.5 | 62.5 | 125 | 250 | 125 |
| 10 | |
81 | 15.625 | 7.8125 | 125 | 125 | 250 |
| 11 | |
74 | 62.5 | 31.25 | 125 | 250 | 250 |
| 12 | |
78 | 31.25 | 15.625 | 125 | 250 | 125 |
| 13 | Ciprofoxacin | - | 15.625 | 15.625 | 15.625 | 15.625 | - |
| 14 | Rifampicin | - | - | - | - | - | 15.625 |
| a Isolated yields | |||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们