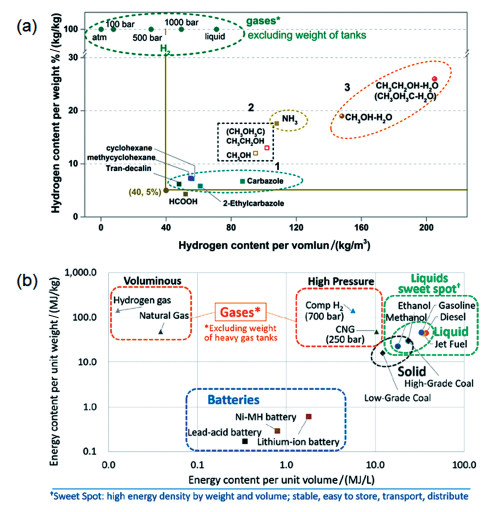
图 1.
各储能介质的性能比较
(a)储氢介质的单位质量储氢和体积储氢密度比较;(b)各类能源的单位质量储能和单位体积储能比较[10]
Figure 1.
Comparison of the performances of energy storage carriers

在过量碳排放导致的全球气候变化日趋严重的大背景下,洁净替代性能源的发展刻不容缓[1]。目前,广受关注的洁净能源包括太阳能和源于太阳能的次生能源,如风能、水能、海洋能、生物质能等[2]。与传统化石能源相比,洁净能源能量密度相对较低,分布受地理环境限制较大,不可避免地需要进一步转化为其他形式的二次能源以便于输送和利用。在众多的二次能源中,氢能简单易得、环境友好。可再生的能源转化而来的氢能本质上是一种“负碳排放”的能源。20世纪70年代以来,“氢能经济”的概念逐渐深入人心,氢能也成为世界各国着力开发的重要新能源方向。
目前限制氢能大规模发展的重大瓶颈是氢能的储存和输送,而该问题的产生与氢气独特的物理化学性质密不可分。一方面,氢气是常见化学物质中的质量能量密度最高的分子,其高位热值达142MJ/kg。但氢气常压下体积密度仅有0.089kg/m3,约为空气密度的1/14,因此其体积能量密度不足12.7MJ/m3,远低于传统油气资源,储运效率低、经济性差。同时,氢气分子半径小,长期安全存储同样是一大技术挑战。因而,为实现氢能高效、安全、经济的应用,开发高效的氢能大规模储运技术至关重要[3~5]。为评价各类氢气储运技术的应用前景,国际能源署估算了实现商业化储氢技术所需的最低单位体积和单位质量储氢密度要求,分别为40kg/m3和5% kgH2/kg[6]。为达到这一标准,分子态存在的H2需要被加压至700bar以上或以低温液态形式加以存储,其中涉及的安全与经济问题一直是储氢技术发展面临的主要挑战。近年来国际储氢技术的基础研究从多维度展开,除了将氢气以分子形式存储的直接储氢外,间接储氢技术—将氢气以化合物形式加以储存也是重要的发展方向[7~9]。其中,有机醇类,特别是甲醇为代表的循环储氢分子具有更高的单位质量和单位体积储氢密度以及良好的化学稳定性; 其氢能储放反应相关的催化和工程技术发展均较为成熟,条件也相对温和,因此成为备受关注的液态氢储存平台分子[10, 11]。诺贝尔奖得主George Olah在“甲醇经济”的构想中将甲醇-H2体系视为后油气时代能源战略的关键[12]。随着世界各国氢能应用的逐步推进,甲醇-H2能源体系相关的化学化工问题将日渐成为基础研究和技术开发的热点。
催化剂的开发与性能的改进是提升甲醇产氢效率、抑制副反应和降低全流程能耗的基础问题。本文将对作为甲醇-H2能源体系基石的甲醇制氢反应催化剂的最新研究进展和机理认识进行综述,讨论甲醇制氢催化剂研发面临的难点与挑战,并对催化剂开发的发展方向加以展望。
白春礼等四位院士在中国科学院的一份题为《液态阳光—全民绿色未来的机遇和途径》的展望文章中[10],明确提出发展液体阳光的策略并充分肯定了将太阳能固定于液态有机分子这一能源发展战略的可行性。其中甲醇作为一种常规条件下的液体燃料被认为是最主要的“液态氢”和“液态电”的载体,可以利用现有的燃料输运基础设施,满足人类在交通、工业和材料等终端应用领域对清洁能源的需求。
储氢介质的单位储氢密度和材料的经济性是衡量氢能载体性能和应用潜力的关键性指标。各种氢能储运介质的质量和体积储氢密度如图 1所示。其中,芳香化合物等可逆储氢分子的储氢质量百分比在5%~7.6%之间,体积密度约45~85kg/m3(图 1,区域1);NH3和甲醇等可循环储氢分子由于可以完全释放分子内的氢,其单位储氢密度显著大于芳香化合物。例如,NH3发生直接脱氢反应(图 1,区域2),理论储氢质量百分比可达17%;甲醇直接脱氢理论储氢率约为12.5%。然而甲醇直接脱氢反应的主产物是CO,会导致下游氢能应用Pt基催化剂中毒失活,因此不能直接加以利用。考虑到下游氢能应用的需求,经过催化重整反应将甲醇分子储存的氢加以释放是更为合理有效的路线。该过程不仅能有效降低制备的燃料气中的CO含量,还同时利用储氢分子的还原能力,进一步从水中取得额外1当量的氢气,从而使甲醇单位质量的储氢密度突破理论上限,进一步提高到18.75%;在类似的逻辑下,二甲醚、乙醇等分子如能完全重整制氢,则可使其单位质量储氢密度进一步提升至26%(图 1,区域3)。醇类极高的质量和体积能量密度表明其是一类理想的储能介质,在高效催化重整过程的辅助下,其储能密度可达各类储能电池的10~50倍,与现有其他化石能源基本持平(图 1(b))[10]。
(a)储氢介质的单位质量储氢和体积储氢密度比较;(b)各类能源的单位质量储能和单位体积储能比较[10]
尽管具有相对更高的理论单位质量储氢密度,可循环储氢分子性能需要高效的催化储氢和放氢反应才能兑现。例如,乙醇分子中含有的C-C键需要高温转化才能实现完全重整产氢,且乙醇重整副产物CO、CH4等选择性较高[13, 14],即现有催化体系无法实现低温高效催化乙醇重整制氢反应,导致乙醇实际单位质量储氢密度远低于理论值。二甲醚与乙醇分子是同分异构体,具有相同的单位储氢密度,且二甲醚相比乙醇重整条件温和。但是二甲醚常温常压下为气体,相比在常温下为液体的甲醇在应用上有一定的不足。在各个循环储氢分子中,甲醇催化重整技术更为成熟可靠,条件更温和,副产物少,效率高,因而受到了广大研究者的青睐。
在氢能应用的构想中,基于可再生能源生产的绿色氢能够存储于氢能载体分子(如甲醇)中,实现高效运输、分配和存储,以供下游的加氢站使用或直接加注于分步式燃料电池系统中构建一体化的“甲醇原位制氢-燃料电池”系统[15]。甲醇直接以燃料的形式加注能够避免加氢站建设的巨大成本投入,并发挥与现有的基础设施联用等优势。除此之外,醇重整与高温燃料电池的联用技术路线也被众多科研机构和企业深度开发(图 2)。
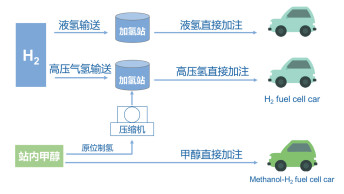
甲醇作为氢能载体在远距离(>200km)输送经济性方面较直接使用氢气具有较强的竞争力。目前已运行的“高压气态氢输送-高压氢直接加注”的技术路线中,经核算其氢气的成本约为60~80元/kg。其中氢气输送成本是其成本偏高的主要原因[16]。与之相比,以年产千兆吨的煤基甲醇为原料,一套规模为1000m3/h的甲醇-蒸汽制氢转化装置制备的氢气成本一般不高于2元/m3,重整制氢的成本约20元/kg左右。综合考虑后续流程中的H2提纯、各项设备折旧、人员费用和利润等各项因素,加氢站终端H2的售价预计约为40~60元/kg。随着可替代清洁能源的发展,利用可再生新能源将CO2转化制备的甲醇有望替代煤基甲醇,从而真正意义上实现碳中性的能源循环利用网络,但该甲醇的成本未见报道。对于氢能应用的主要终端,氢燃料电池汽车的百公里能源消耗约为1kg H2/100km[17]。按前述氢气价格计算,百公里燃料成本约60元;目前燃油乘用车百公里能源消耗约为8L/100km,按最新油价大概7元/L计算,百公里成本为56元;所以甲醇氢燃料电池汽车与燃料车在能源消耗消费层面大体持平。但是燃油车百公里消耗的总热值为255.2MJ,CO2排放量经核算约为18.35kg/100km;对于甲醇氢燃料电池来说,百公里总热值仅需124MJ,相应的碳排放也仅为7.3kg/100km(表 1)。考虑到氢能汽车在CO2减排层面的优良表现(较燃油车CO2减排约60%),有助于实现我国在气候变化巴黎大会上“2030年单位国内生产总值二氧化碳排放比2005年下降60%~65%”的承诺,其推广应用可以获得显著的社会和环境效益。而且随着氢能的普及以及相关政策法规的完善,甲醇制氢体系较传统燃油车的竞争力还有望进一步提升。
 下载:
导出CSV
下载:
导出CSV
| 项目 | 燃油车 | 甲醇氢汽车 |
| 100km耗能 | 8L | 1kg |
| 单价 | ~7.0元/L* | 60元/kg |
| 总价/元 | 56 | 60 |
| 单位热值 | 31.9MJ/L | 124MJ/kg |
| 总热值/MJ | 255.2 | 124 |
| CO2排放量/kg | 18.35 | 7.3 |
| * 2020-07-20燃油车汽油92#,价格按全国油价均价计算 | ||
在不远的将来,使用甲醇替代分子氢有望构建更加经济便捷安全清洁的氢气供应消费网络。其中决定甲醇-氢能源体系成功与否的关键反应之一是甲醇催化产氢。
从分子层面分析,甲醇产氢反应的本质是将分子内的全部氢原子释放的过程,这其中主要涉及的化学变化包括C-H、O-H键等化学键的解离以及碳原子从低价经多步反应氧化为CO2。在甲醇制氢反应中氧化剂的选择对制氢反应的热力学、产氢效率和反应器的设计优化和反应条件均会产生显著的影响。水和分子氧是最常见的氧化剂。根据引入氧化剂的特点,目前广为研究的甲醇制氢反应主要分为以下几种实现形式:甲醇水蒸气重整(SRM)[18~20]、甲醇氧化重整(OMR)[21, 22]和甲醇部分氧化(POM)[23, 24]等(表 2)[25]。
下载:
导出CSV
| 序号 | 反应类型 | 反应式 | 优势 | 劣势 | 技术成熟度 |
| 1 | SRM | CH3OH(g)+H2O(g)=CO2+3H2;ΔH=49.7kJ/mol | 产氢量高(75%) | 较耗能,启动慢 | 成熟 |
2 |
OMR | CH3OH(g)+x/(x+y)H2O(g)+y/2(x+y)O2= CO2+(3x+2y)/(x+y)H2;ΔHo=(49x-192y)/ (x+y)kJ/mol |
易于启动、反应迅速 | 出口氢浓度(41%~70%)*、 操作复杂 |
开发中 |
| 3 | POM | CH3OH(g) + 1/2 O2=CO2 + 2H2; ΔHo=-192kJ/mol |
易于启动、反应迅速 | 出口氢浓度低(41%)*、 存在热点致催化剂失活 |
开发中 |
| *O2直接来源于空气,生成的H2浓度会被空气中大量的N2稀释;序列2、3中的出口氢浓度是按空气稀释进行折算的 | |||||
根据化学计量关系,甲醇与水反应的重整制氢过程(SRM)能在释放甲醇分子内全部氢的同时实现水中取氢,并获得额外1分子甲醇当量的氢气。重整气中氢气浓度在三类制氢方法中最高(75%)(表 2,序列1)。但是甲醇-水重整制氢过程在热力学上是一个高温有利的吸热反应(ΔH=49.7kJ/mol),目前实际应用和基础研究中报道的甲醇-水重整制氢过程的工作温度一般高于250℃。相对较高的工作温度和汽化单元的存在导致分布式甲醇制氢系统在启动工况下的响应较慢。然而,对于连续现场制氢、现制现用的工业化应用来说,如作为加氢站氢气来源的前端(图 2,路径3),SRM制氢技术的H2含量高、技术成熟,是当前制氢反应的最佳选择。
以氧气部分或完全替代水作为氧化剂可以显著改变甲醇制氢反应的反应热力学。当反应气氛中分子氧的含量超过水浓度的1/8时,甲醇制氢反应即转化为放热反应。利用这一方式开发的空气-水-甲醇共进料的制氢过程被称为甲醇氧化重整,或甲醇自热重整(OMR);如完全使用空气作为氧化剂,则反应称为POM制氢。上述过程在实际体系中响应较快,大幅提升能源利用效率,减少附加装置的配备,简化工艺流程。根据表 2中化学反应计量关系,自热重整过程中每分子甲醇能产生2~3分子氢。由于氧化重整是以空气为氧化剂,每分子氧气的消耗就会引入1.88当量的N2,导致出口氢气的浓度在41%~70%。对于POM制氢来说,每分子甲醇仅能获得2分子氢,实际出口氢气的浓度仅为41%。在甲醇制氢中引入氧化剂,虽然制氢能耗降低,但是氢气选择性的控制较水蒸气重整难度大幅提高,易出现过度氧化的产物;另外空气作为氧化剂,也可能导致氮氧化物等环境污染物生成;同时氧化放热反应对反应器换热要求较高,催化剂容易在局部热点的影响下烧结失活。OMR或POM制氢的技术还处在开发中,尚未实现产业化。
经过比较可知,在多种甲醇制氢方式中甲醇-水重整制氢反应产氢率高、选择性控制简便,是目前催化剂合成和工艺开发较为成熟的领域[27]。从工程层面,甲醇重整前期启动所需要的能量可以通过耦合小型储能电池的方式加以解决[26, 28]。因此,接下来将主要针对甲醇重整制氢催化剂的研究进展和面临的挑战进行进一步的阐述。
甲醇-水重整制氢催化剂的开发和改进对分布式加氢站现场制氢模式的推广具有重要的推动作用。目前,商业化Cu/ZnO/Al2O3催化剂在SRM制氢应用中得到了广泛的认可,是目前应用的主流。催化剂的助剂、载体酸碱性等改性策略的研究也大幅度提升了Cu基催化剂在SRM制氢中的催化活性和稳定性[29, 30]。商用SRM产氢技术能够在连续工作状态下实现在10~10000 Nm3/h规模内的产氢,产能灵活可调。Cu基催化剂面临的主要问题是其在间断的启停状态下稳定性不够理想,尤其在水蒸气凝结状况下极易失活[31, 32]。为此科学家也针对性地研究了以铂族贵金属为活性中心的负载型催化剂,以提高催化剂的稳定性。然而,受贵金属本征催化性质的影响,贵金属甲醇重整催化剂上C-H、C-O键解离速率相对偏高,甲醇分解、氢解、甲烷化等副反应选择性高,制备的氢气中CO、甲烷等副产物含量远高于传统铜基催化剂。Sá等[31]按Cu基催化剂和铂族贵金属催化剂的SRM的催化剂进行了详尽的归类和催化性能列举;Palo等[8]不仅对甲醇重整的催化剂技术进行了阐述,同时对重整反应器的设计、甲醇重整制氢-燃料电池氢能应用技术、其他储氢材料-燃料电池氢能应用技术与甲醇直接燃料电池等技术路线的优劣势进行了对比剖析。
鉴于SRM已有的诸多综述报道[3, 8, 15, 31],本文将主要关注近期新兴的甲醇-水液相重整制氢体系的优势、催化剂的设计和开发以及催化机理。
醇类-水液相重整产氢是Cortright等[33]于2002年首次提出,在该过程中反应物不经汽化,直接以液态的形式发生重整反应产氢。与传统水蒸气重整反应相比,液相重整反应减少了反应物汽化的步骤,流程更为紧凑,能耗较低(图 3)。同时,在液相反应条件下,产物中残留的CO的浓度较水蒸气重整大大降低,有望在后续H2纯化步骤中精简水煤气变换或甲烷化氢气净化提纯装置,直接通过CO选择性氧化或Pd膜反应器等手段联用获得高纯氢,是一种广受关注的醇类制氢新体系[34]。
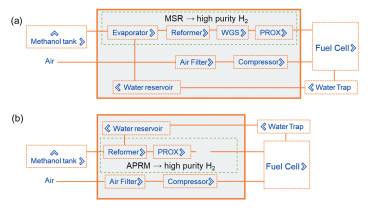
(a)甲醇水汽相重整-氢燃料电池[26];(b)甲醇-水液相重整-氢燃料电池
Cortright等比较了甲醇、乙二醇、甘油、葡萄糖等生物质基醇类的液相催化重整产氢行为。以3%Pt/Al2O3为催化剂在220℃条件下反应,甲醇-水液相重整产氢的速率为4×104μmol·g-1·h-1,氢气的选择性为99%[33]。该催化剂在反应温度相同的水蒸气重整固定床反应器中,副产物CO的选择性高达70%,充分说明在液相反应条件下更有利于CO发生水煤气变换,实现水中取氢。Sn修饰非贵金属Raney-Ni在液相重整中表现出和贵金属Pt接近的催化活性和选择性,在Sn/Ni的原子比为1∶14时表现出最优的催化活性[35]。研究者对催化剂载体的酸性、金属中心对重整的影响也进行了广泛的对比研究,发现酸性载体和酸性溶液有利于重整中烃类的生成,降低了H2的收率;Pt、Pd、Ni、Sn等金属中心有利于重整产氢反应的发生[36, 37],而Ru、Rh等金属倾向于解离醇类C-O键,利于烷烃的生成,Mo、Fe等助剂则能有效提升Pt/Al2O3催化剂的重整产氢活性[38]。在此基础上,Miyao等[39]对PtRu双金属甲醇水液相重整活性的载体效应进行了评价,发现在SiO2、Al2O3、TiO2、MgO、CeO2和ZrO2等载体中,TiO2负载的PtRu双金属合金在80℃表现出最高的活性和选择性(nCO2/n(CO2+CO)>90%)。Park等[40]发现,TiO2载体上均匀分散的MoOx纳米团簇能够显著提升Pt基催化剂的活性,在190℃下产氢速率达800h-1,副产物选择性CO/CO2和CH4/CO2均小于1%。在压力变化对反应性能影响的研究中发现,Pt-MoOx/TiO2和Cu/ZnO/Al2O3催化剂的产氢活性与压力表现出反相关,这个结果与其他报道的Pt基催化剂不同,其具体成因仍需进一步研究加以解释。
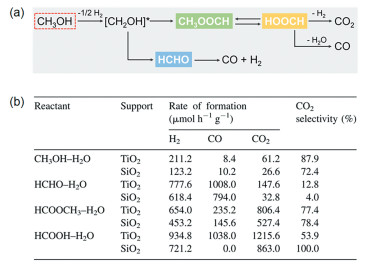
研究者观测到甲醇在水汽重整产氢过程中的中间物种主要为甲醛、甲酸甲酯或甲酸三类(图 4(a))。由于高温液相红外原位检测催化剂表面物种存在技术困难,Miyao等根据水汽重整的机理认识,将可能的中间物种甲醛、甲酸甲酯或甲酸分别作为液相重整的反应物来进行反应[39]。将对应反应的产物选择性与PtRu/TiO2催化剂催化甲醇水液相重整的选择性进行比较(图 4(b))发现,以甲酸甲酯为反应物时,产物中CO2的选择性与甲醇为反应物时最为相近(~80%);而以甲酸和甲醛为中间物种时,CO2的选择性分别仅为54%和12%,远远偏离原始反应的催化选择性。由此他们推测甲酸甲酯是PtRu/TiO2催化剂在甲醇水液相重整反应过程的主要中间物种。然而,研究者在讨论中忽略了甲醇可以经过催化脱氢形成中间物种CO,而CO再经水汽迁移反应(CO+H2O→CO2+H2)形成CO2的反应路径。
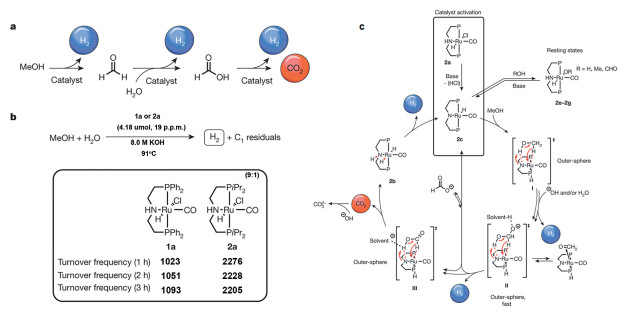
均相催化剂也可以用于催化甲醇产氢反应[41~45],Nielsen等[41]开发了由三苯基膦胺基等有机配体稳定的单核Ru均相催化剂,在91℃时催化剂的产氢速率达到2668h-1,是目前报道的90℃下甲醇产氢反应的最高产氢速率。然而,该产氢过程并非严格的甲醇-水重整,而是采用高浓度的氢氧化钠作为甲醇脱氢的氧化剂,反应式可以写为CH3OH+2NaOH=3H2+Na2CO3,即每产生3分子的氢气就需消耗2分子的氢氧化钠,即反应介质中的碱是甲醇产氢的牺牲剂。因此,在甲醇脱氢过程中氢氧化钠的浓度会发生持续改变,随着碱浓度的降低,催化剂活性会发生断崖式下降。因此,虽然Ru配合物催化剂在100℃以下实现了高效产氢,能够与低温质子燃料电池在温度上完美匹配,但是反应体系所需的高碱性环境以及碱的消耗是均相催化剂在后续应用推广中面临的难题。
机理研究充分揭示了碱性试剂对于反应的关键作用[41, 46]。在配合物中,Ru金属中心是催化甲醇分子C-H键断裂的活性中心,而三苯基膦胺基配体是甲醇分子中-OH键或溶剂中-OH键的活化中心。从反应机理图中可发现,在形成甲醛中间物种时,为了有效抑制中间物种甲醛的分解形成副产物CO,溶液中的羟基物种对C端的进攻是必不可少的;同时羟基也是在温和条件下移除N原子上的H,再生O-H键解离中心的关键。溶液中高浓度的NaOH提高了反应介质中OH-的浓度,极大促进了Ru有机配合物催化反应速率。金属Pincer类配合物高效的甲醇制氢性能主要源于其配合物独特的双中心结构以及金属Ru-N双功能中心之间合适的几何距离和空间结构(图 5)。
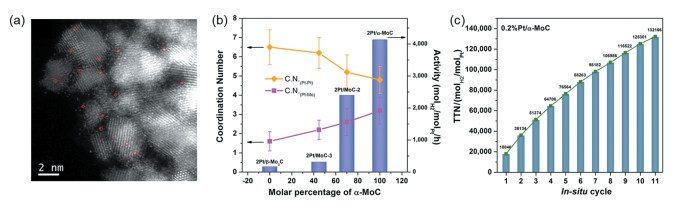
单原子催化剂是近年来提出和发展的一类新型催化体系。具有多相催化剂便于分离再生的固有优势,又能够达到传统均相催化剂和酶催化剂才能实现的百分百金属原子利用率。基于对传统甲醇液相重整反应多相与均相催化剂的理解,笔者课题组[47]以原子级高度分散的Pt模拟配合物中的金属中心,以新型立方相α-MoC为载体构建了双功能的负载单原子催化剂(图 6(a))。Pt/α-MoC在甲醇水液相重整反应出表现出超高的产氢活性,且无需碱性溶液作为助剂,在190℃下其单位Pt中心产氢活性高达18064h-1(图 6(c))。原子级分散的金属中心铂完全暴露在表面,使活性位密度最大化,进一步提升催化剂的产氢效率。该催化剂上CO副产物的选择性 < 1%,对后续氢气的分离提纯要求较低,表现出了一定的应用前景。扩展X-射线吸收精细谱证明Pt-Mo配位数达2.6(图 6(b)),说明在还原性气氛甲烷-氢的作用下,铂与载体α-MoC会形成化学键,是二者之间存在载体金属强相互作用的表现。原位XPS和X射线近边吸收光谱(XANES)表征显示Pt/α-MoC催化剂上Pt表现出极强的正电性,表明强相互作用会诱导电子转移,使Pt的电子密度降低,更有利于重整反应。
Pt/α-MoC催化剂对甲醇-水重整反应的机理研究主要是通过程序升温表面反应(TPSR)方法与理论计算相结合完成的(图 7)。Pt/α-MoC催化剂的高效产氢活性和高选择性与其微观反应路径密不可分。通过甲醇-水在α-MoC表面的TPSR实验,可以发现α-MoC具有优异的水和甲醇中-OH键的解离活性(166℃),因为此时H2的生成未伴随含碳物种的生成;而CO2产物只在更高的反应温度下才被检出,说明甲醇中C-H键的断裂在α-MoC上能垒远高于O-H键解离。与之相比,Pt/α-MoC的TPSR实验表明甲醇重整反应在115℃时即发生,大量H2的生成伴随着CO2的产生,说明原子级分散的Pt是甲醇分子中C-H键活化的中心。对比Pt/α-MoC甲醇TPD的实验现象,可以推测水分子在Pt/α-MoC体系中对重整制氢反应的发生具有重要的推动作用。结合对照组Pt/Al2O3 TPSR的实验结果,我们可发现如果载体没有优异解离水能力用以促进水煤气变换反应的发生,则甲醇的主要反应路径是分解形成CO和氢气。TPSR实验结果说明了Pt/α-MoC具有类似于Ru有机配合物催化剂的双功能性质,Pt-α-MoC的协同作用促进了甲醇-水重整制氢反应的高效进行(图 7a~d)。
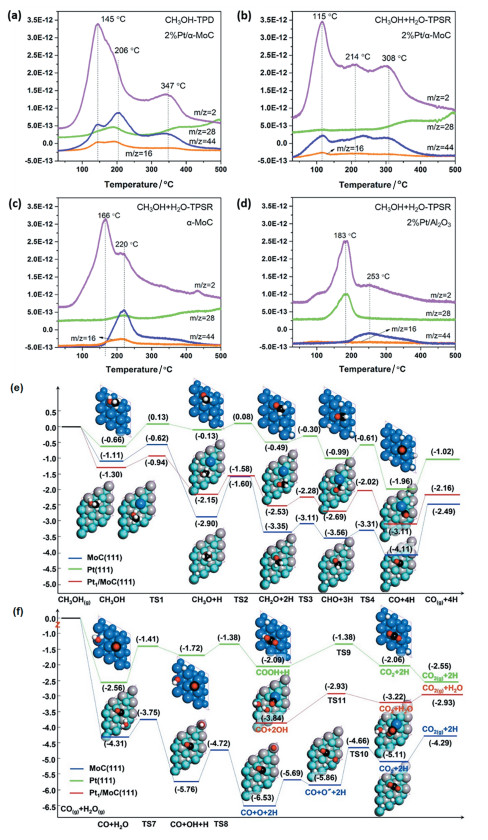
在此认识基础上构建催化剂模型再结合理论计算,可从原子角度更深层次理解催化反应路径。理论计算证实了甲醇分子主要在原子级分散的金属中心铂原子上发生C-H键断裂(0.57eV),且形成吸附态的Pt-CO;载体α-MoC解离水至OH的能垒仅为0.56eV,出色的低温解离水能力促进表面形成高覆盖度的OH。甲醇分解至CO、水解离形成OH两个基元步骤极低的能垒和高度匹配的速率促进了Pt-Mo界面处高效水煤气变换反应的发生,生成CO2和H2释放催化活性位(图 7e~f)。
载体α-MoC优异的水活化性能也可以用于甲醇水蒸气重整催化剂的改进。最近Cai等[48]报道,Pt-Zn/α-MoC在甲醇水蒸气重整制氢反应中同样可以表现出良好的产氢速率和极低的CO杂质选择性,证明了通过强化水的活化能力提升产氢催化剂性能思路的可行性。
世界能源发展正处在历史第三次能源革命的十字路口—油气能源向新能源的转换期。能源类型表现出从高碳向低碳、非碳方向转化的发展趋势。“甲醇水重整制氢”的甲醇-氢能源体系表现出良好的经济性,有望解决氢能应用中储存和输送的瓶颈问题。随着甲醇制氢技术的进步和催化剂的进一步发展迭代,其作为加氢站氢源的优势将日益显现,能够助力节能减排目标的实现与生态环境的构建。
“甲醇制氢-氢燃料电池”一体化的甲醇氢燃料电池汽车是氢能进一步发展的目标,醇氢燃料电池的发展可以免去高昂的加氢站等基础设施建设成本,与现有的油气体基础设施实现共用,真正的将“加气”变成“加油”,助力氢能源的推广。现阶段,若干醇氢概念车型已经发布,充分证明了该思路的可行性。作为技术开发的基石,低温甲醇催化重整制氢反应催化剂的研发和基础科学问题的解决是进一步提升甲醇-氢动力系统效能的关键。在性能开发层面,进一步提高催化剂活性,达到能够在有限的空间和催化剂质量下产出满足车载燃料电池系统消耗氢气的指标是实现商用的基本要求。
除此之外,实现在多次的“启动-停止”以及在相对复杂工况下催化剂高度的稳定性、产氢条件温和化、降低催化成本和简化催化剂再生条件等都是亟待解决的关键科学问题。目前,基于双功能催化中心耦合强化C-H、O-H键活化反应的催化剂开发策略已被均相催化和原子级分散催化剂的成功所证明。进一步通过催化剂的筛选和理论预测寻找适合的甲醇制氢反应的双功能催化材料组合将成为下阶段研究的重点。相信随着催化剂开发的逐渐深入,甲醇-水液相重整与燃料电池联用的动力组合将为氢能汽车的发展提供新的思路和方向。
Obama B. Science, 2017, 355(6321): 126~129. doi: 10.1126/science.aam6284
Kamat P V. J. Phys. Chem. C, 2007, 111(7): 2834~2860. doi: 10.1021/jp066952u
Deng Z Y, José M F F, Sakka Y. J. Am. Ceram. Soc., 2008, 91(12): 3825~3834. doi: 10.1111/j.1551-2916.2008.02800.x
Aaldto-Saksa P T, Cook C, Kiviahob J, et al. J. Power Sources, 2018, 396: 803~823. doi: 10.1016/j.jpowsour.2018.04.011
Schlapbach L, Zuttel A. Nature, 2001, 414(6861): 353~358. doi: 10.1038/35104634
Teichmann D, Arlt W, Wasserscheid P, et al. Energy Environ. Sci., 2011, 4(8): 2767~2773. doi: 10.1039/c1ee01454d
Palo D R, Dagle R A, Holladay J D. Chem. Rev., 2007, 107(10): 3992~4021. doi: 10.1021/cr050198b
Xie Y J, Hu P, Ben-David Y, et al. Angew. Chem. Int. Ed., 2019, 58(15): 5105~5109. doi: 10.1002/anie.201901695
Shih C F, Zhang T, Li J H, et al. Joule, 2018, 2(10): 1925~1949. doi: 10.1016/j.joule.2018.08.016
Chen S, Pei C L, Gong J L. Energy Environ. Sci., 2019, 12(12): 3473~3495. doi: 10.1039/C9EE02808K
Olah G A. Angew. Chem. Int. Ed., 2005, 44(18): 2636~2639. doi: 10.1002/anie.200462121
Miyamoto Y, Akiyama M, Nagai M. Catal. Today, 2009, 146(1-2): 87~95. doi: 10.1016/j.cattod.2008.12.033
Hou T F, Zhang S Y, Chen Y D, et al. Renew. Sust. Energ. Rev., 2015, 44: 132~148. doi: 10.1016/j.rser.2014.12.023
Iulianelli A, Ribeirinha P, Mendes A, et al. Renew. Sust. Energ. Rev., 2014, 29: 355~368. doi: 10.1016/j.rser.2013.08.032
司伟, 于栋. 证券研究报告, 2019.
Yu K M, Tong W Y, Adam W, et al. Nat. Commun., 2012, 3: 1230. doi: 10.1038/ncomms2242
Setthapun W, Bej S K, Thompson L T. Top. Catal., 2008, 49(1-2): 73~80. doi: 10.1007/s11244-008-9070-7
Oihane S, Ion V, Iñigo P. Int. J. Hydrogen Energy, 2016, 41: 5250~5259. doi: 10.1016/j.ijhydene.2016.01.084
Pojanavaraphan C, Nakaranuwattana W, Luengnaruemitchai A, et al. Chem. Eng. J., 2014, 240: 99~108. doi: 10.1016/j.cej.2013.11.062
Liu S T, Takahashi K, Fuchigami K, et al. Appl. Catal. A, 2006, 299: 58~65. doi: 10.1016/j.apcata.2005.10.012
Wang Z F, Xi J Y, Wang W P, et al. J. Mol. Catal. A, 2003, 191(1): 123~134. doi: 10.1016/S1381-1169(02)00352-7
Hernández-Ramírez E, Wang J A, Chen L F. Appl. Surf. Sci., 399: 77~85.
Yong S T, Ooi C W, Chai S P, et al. Int. J. Hydrogen Energy, 2013, 38(22): 9541~9552. doi: 10.1016/j.ijhydene.2013.03.023
Chen W H, Lin B J. Int. J. Hydrogen Energy, 2013, 38(24): 9973~9983. doi: 10.1016/j.ijhydene.2013.05.111
Araya S S, Liso V, Cui X T, et al. Energies, 2020, 13(3): 596. doi: 10.3390/en13030596
Pan L W, Wang S D. Int. J. Hydrogen Energy, 2005, 30(9): 973~979. doi: 10.1016/j.ijhydene.2004.10.012
Choi Y, Stenger H G. Appl. Catal. B, 2002, 38(4): 259~269. doi: 10.1016/S0926-3373(02)00054-1
Agrell J, Birgersson H, Boutonnet M, et al. J. Catal., 2003, 219(2): 389~403. doi: 10.1016/S0021-9517(03)00221-5
Sá S, Silva H, Brandão L, et al. Appl. Catal. B, 2010, 99(1-2): 43~57. doi: 10.1016/j.apcatb.2010.06.015
Twigg M V, Spencer M S. Top. Catal., 2003, 22(3-4): 191~203.
Cortright R D, Davda R R, Dumesic J A, et al. Nature, 2002, 418(6901): 964~967. doi: 10.1038/nature01009
Davda R R, Shabaker J W, Huber G W, et al. Appl. Catal. B, 2005, 56(1-2): 171~186. doi: 10.1016/j.apcatb.2004.04.027
Huber G W, Shabaker J W, Dumesic J A, et al. Science, 2003, 300(5628): 2075~2077. doi: 10.1126/science.1085597
Li D D, Li Y, Liu X H, et al. ACS Catal., 2019, 9(10): 9671~9682. doi: 10.1021/acscatal.9b02243
Coronado I, Stekrova M, Moreno L G, et al. Biomass Bioenergy, 2017, 106: 29~37. doi: 10.1016/j.biombioe.2017.08.018
Sakamoto T, Morishima H, Yoshida A, et al. Catal. Lett., 2009, 131(3-4): 419~424. doi: 10.1007/s10562-009-0098-5
Miyao T, Yamauchi M, Narita H, et al. Appl. Catal. A, 2006, 299: 285~291. doi: 10.1016/j.apcata.2005.10.043
Park J H, Kim Y T, Park E D, et al. ChemCatChem, 2013, 5(3): 806~814. doi: 10.1002/cctc.201200458
Nielsen M, Alberico E, Baumann W, et al. Nature, 2013, 495(7439): 85~89. doi: 10.1038/nature11891
Monney A, Barsch E, Sponholz P, et al. Chem. Commun., 2014, 50(6): 707~709. doi: 10.1039/C3CC47306F
Alberico E, Sponholz P, Cordes C, et al. Angew. Chem. Int. Ed., 2013, 52(52): 14162~14166. doi: 10.1002/anie.201307224
Prichatz C, Alberico E, Baumann W, et al. ChemCatChem, 2017, 9(11): 1891~1896. doi: 10.1002/cctc.201700015
Andérez-Fernández M, Vogt L K, Fischer S, et al. Angew. Chem., 2017, 129(2): 574~577. doi: 10.1002/ange.201610182
Alberico E, Lennox A J J, Vogt L K, et al. J. Am. Chem. Soc., 2016, 138(45): 14890~14904. doi: 10.1021/jacs.6b05692
Lin L L, Zhou W, Gao R, et al. Nature, 2017, 544(7648): 80~83. doi: 10.1038/nature21672
Cai F F, Ibrahim J J, Fu Y, et al. Appl. Catal. B, 2020, 264: 118500. doi: 10.1016/j.apcatb.2019.118500

图 1 各储能介质的性能比较
Figure 1 Comparison of the performances of energy storage carriers
(a)储氢介质的单位质量储氢和体积储氢密度比较;(b)各类能源的单位质量储能和单位体积储能比较[10]

图 2 氢气与甲醇-H2能源体系的不同应用形式
Figure 2 The illustrative scheme of the applications of hydrogen and methanol-H2 energy system

图 3 甲醇-氢燃料电池一体化
Figure 3 Illustration of the integration of methanol reformer with H2 fuel cell
(a)甲醇水汽相重整-氢燃料电池[26];(b)甲醇-水液相重整-氢燃料电池




表 1 甲醇氢燃料电池汽车与燃油车的经济性和碳排放比较
Table 1. Comparison of economy and carbon emissions between methanol-H2 fuel car and gasoline vehicles
| 项目 | 燃油车 | 甲醇氢汽车 |
| 100km耗能 | 8L | 1kg |
| 单价 | ~7.0元/L* | 60元/kg |
| 总价/元 | 56 | 60 |
| 单位热值 | 31.9MJ/L | 124MJ/kg |
| 总热值/MJ | 255.2 | 124 |
| CO2排放量/kg | 18.35 | 7.3 |
| * 2020-07-20燃油车汽油92#,价格按全国油价均价计算 | ||
 下载: 导出CSV
下载: 导出CSV
| 序号 | 反应类型 | 反应式 | 优势 | 劣势 | 技术成熟度 |
| 1 | SRM | CH3OH(g)+H2O(g)=CO2+3H2;ΔH=49.7kJ/mol | 产氢量高(75%) | 较耗能,启动慢 | 成熟 |
2 |
OMR | CH3OH(g)+x/(x+y)H2O(g)+y/2(x+y)O2= CO2+(3x+2y)/(x+y)H2;ΔHo=(49x-192y)/ (x+y)kJ/mol |
易于启动、反应迅速 | 出口氢浓度(41%~70%)*、 操作复杂 |
开发中 |
| 3 | POM | CH3OH(g) + 1/2 O2=CO2 + 2H2; ΔHo=-192kJ/mol |
易于启动、反应迅速 | 出口氢浓度低(41%)*、 存在热点致催化剂失活 |
开发中 |
| *O2直接来源于空气,生成的H2浓度会被空气中大量的N2稀释;序列2、3中的出口氢浓度是按空气稀释进行折算的 | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们