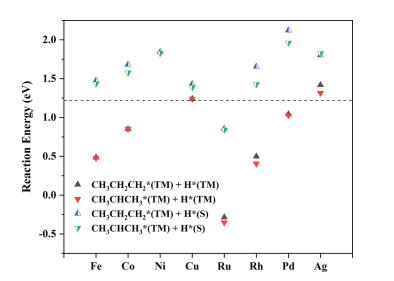
Figure 1.
The reaction energies of the first dehydrogenations of propane on TM1/MoS2. The dashed horizontal line (potential energy = 1.22 eV) is selected to indicate the corresponding value of the Pt-Sn catalyst in ref. [32].
A mechanistic study of selective propane dehydrogenations on MoS2 supported single Fe atoms
Yingke Yang , Ruru Song , Xing Fan , Yunxia Liu , Ningning Kong , Haiping Lin , Youyong Li

Light olefins are employed as feedstock in the production of a vast array of chemicals, including polymers (e.g., polyethylene and polypropylene), oxygenates (e.g., ethylene glycol, acetone and propylene oxide), and important chemical intermediates (e.g., ethylbenzene and propionaldehyde) [1]. Propylene is the second largest-produced petrochemical with a current global production of approximately 100 million tons (Mt) per annum and is expected to increase steadily at an annual rate of 3%~4% [2]. Traditionally, propylene is obtained through steam cracking and fluid catalytic cracking (FCC) of oil refineries, accounting for 90% of the propylene market until 2012 [3]. On top of this, the demand for propylene is expected to grow from 109 million tons in 2014 to about 165 million tons by 2030, roughly 12%~14% higher than the total production capacity of conventional technologies [4]. Nevertheless, the yield of propylene by conventional technologies is rather limited (13 wt%~17 wt%) and coupled to that of ethylene [5]. These have prompted petrochemical industries to seek alternative feedstocks and on-purpose propylene technologies to meet the demand for propylene, such as abundant shale gas, on-purpose propane dehydrogenation (PDH) and methanol-to-propylene (MTP) [6, 7]. In particular, PDH is regarded as one of the most promising methods of propylene production because it can obtain high-purity propylene products rather than mixtures [8].
At present, the challenges of PDH chiefly originate in two aspects: (1) A highly active catalyst is required to cleave sp3C−H bonds in propane (4.31 eV) [9, 10]; (2) C−H bonds in propylene (3.34 eV) are weaker than those in propane, suggesting that over-dehydrogenations and coke formation are faster than propane dehydrogenations [9-11]. In this field, Pt catalysts are very active in C−H bond cleavages but suffer from poor propylene selectivity and fast deactivation caused by coke formation and catalyst sintering at high temperatures [12, 13]. With the continuous improvements of Pt catalysts, the schemes of alloying Pt with a second metal (Sn or Cu) have confirmed that dispersed Pt atoms can activate C−H bonds and the second metal may weaken propylene adsorption and prevent coking [14-16]. Inspired by such a catalytic mechanism, single-atom catalysts (SACs) have been designed and investigated as PDH catalysts. For example, Sun et al. predicted that single Pt atoms supported on the nitrogen vacancy of boron nitride (Pt-Nvac/BN) may be an effective PDH catalyst with a balanced ability to activate C−H bonds in propane and accelerate propylene desorption [17]. Kong et al. revealed that single V atoms anchored on graphitic carbon nitride (V1/g-C3N4) may serve as a promising catalyst for nonoxidative PDH with industrially practical activity, selectivity, and thermal stability [18]. Based on previous studies, more catalytic systems need to be proposed and investigated to optimize the design strategies of SACs for PDH. In the present work, we demonstrate that MoS2 supported single non-noble transition metal (TM) atoms can be employed as inexpensive and efficient PDH catalysts because of highly exposed and unsaturated d orbitals that can promote C−H bond cleavages [19-22] and propylene desorption prior to dehydrogenation [23].
A practical challenge in designing SACs for PDH is that single TM atoms must be firmly anchored on the supporting materials at high temperatures (770~980 K) required by profitable propane conversion efficiency [24, 25]. Recently, Zhao et al. reported a universal seeding strategy (> 800 ℃) to synthesize SACs on different 2D materials, such as graphene, boron nitride (BN) and molybdenum disulfide (MoS2), implying that these SACs (such as Fe1/MoS2, Co1/MoS2, Ni1/MoS2, Cu1/MoS2, Pd1/MoS2, Ag1/MoS2) may maintain thermal stability at high temperatures. In addition, SACs (Fe1/MoS2, Co1/MoS2, Ni1/MoS2, Ru1/MoS2, Rh1/MoS2) have been synthesized via a facile and scalable hydrothermal grafting method at low temperatures (160~180 ℃) [26, 27].
Herein, the PDH catalytic performances of TM1/MoS2 (TM = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag) experimentally reported are investigated by means of density functional theory (DFT) calculations. We find that the first C−H bond cleavage (rate-limiting step) can proceed with a low activation barrier of 0.85 eV on Fe1/MoS2. Although propylene desorption is relatively slow due to a moderate binding energy (1.48 eV), propylene selectivity can be assured by kinetically inhibiting deep dehydrogenations. This work may therefore have crucial influences on developing new SACs for PDH.
Firstly, the adsorption sites and binding energies of single TM atoms on MoS2 are studied. As shown in Fig. S1 (Supporting information), single TM atoms prefer to bind above the Mo-top sites, which is consistent with previous experimental observations [26, 27]. The catalytic performance of PDH can be evaluated by propane activation and propylene selectivity [23]. Based on the Brønsted-Evans-Polanyi relationship, the catalytic activity of PDH can be predicted by the reaction energy (ΔE) of the first C−H bond cleavage (rate-limiting step) [28, 29]. Meanwhile, the propylene selectivity can be estimated by the activation energy difference between dehydrogenation and desorption of propylene. These have been extensively selected as theoretical descriptors to screen promising candidates for PDH catalysts [30, 31]. Mechanistically, PDH is considered to follow the reverse Horiuti-Polani mechanism [31-33], in which propylene and H2 are produced via two successive dehydrogenations of propane [25] along with various side reactions (Scheme S1 and Table S1 in Supporting information).
Before PDH, we explored the different adsorption modes of propane on TM1/MoS2. Here, the primary C atom and the secondary C atom are named Cα and Cβ, respectively. The H atoms carried by Cα and Cβ are named Hα and Hβ, respectively. The adsorption states of Hα atoms and Hβ atoms pointing towards single TM atoms are named the L mode and V mode, respectively (Fig. S2 in Supporting information). Both the L mode and V mode of propane adsorption on TM1/MoS2 are favorable, which may be a good start for PDH.
After the first dehydrogenation of propane, 1-propyl and 2-propyl are adsorbed on single TM atoms as reaction intermediates, and the dissociated H atoms may be either coadsorbed on single TM atoms or captured by the adjacent S atoms (Fig. S3 in Supporting information), which named CH3CH2CH2*(TM) + H*(TM), CH3CHCH3*(TM) + H*(TM), CH3CH2CH2*(TM) + H*(S) and CH3CHCH3*(TM) + H*(S). Interestingly, the reaction energies of the first dehydrogenations on Fe1/MoS2, Co1/MoS2, Ru1/MoS2, Rh1/MoS2 and Pd1/MoS2 (−0.35~1.04 eV) are lower than that of the commercial Pt-Sn catalyst (1.22 eV) as shown in Fig. 1 and Table S2 (Supporting information) [32]. For example, Fe1/MoS2 not only shows lower reaction energies of the first dehydrogenations of propane (0.49 eV and 0.48 eV) but also activates the second C−H bonds (Cβ−Hβ and Cα−Hα), which are elongated from 1.10 Å to 1.22 Å and 1.21 Å, respectively (Fig. S4 in Supporting information).
The dehydrogenations of propane are reversible [24], in which forward and backward reactions proceed through the same transition states and intermediate states [33]. Here, the energy barriers of the forward and reverse reactions are defined as Ef and Er, respectively. As shown in Table S3 (Supporting information), 1-propyl and 2-propyl adsorbed on Co1/MoS2 and Pd1/MoS2 are more favorable for hydrogenation to propane because of low hydrogenation barriers (Er ≈ 0.02~0.09 eV), while propylene is more favorable for hydrogenation to 1-propyl and 2-propyl on Ru1/MoS2 (Er ≈ 0.02~0.07 eV), suggesting the low activity of Co1/MoS2, Pd1/MoS2 and Ru1/MoS2 for PDH. Consequently, propylene may be obtained thermodynamically on Fe1/MoS2 and Rh1/MoS2 by two successive dehydrogenations via reactions 1, 4 (CH3CH2CH3* → CH3CH2CH2* + H* → CH3CH=CH2* + H2) and reactions 2, 5 (CH3CH2CH3* → CH3CHCH3* + H* → CH3CH=CH2* + H2).
After two successive dehydrogenations of propane on Fe1/MoS2 and Rh1/MoS2, the distances between the two dissociated H atoms are only 0.81~0.87 Å (Fig. S5 in Supporting information) and the combination of these two dissociated H atoms to produce a H2 molecule only need to overcome a low barrier that is less than 0.10 eV. We further explored the adsorption and dehydrogenation of propylene on TM1/MoS2. The Cα and Cβ of propylene are bonded to single TM atoms of TM1/MoS2, and the configurations are in good agreement with the "propylene-π" mode (Fig. S5 in Supporting information) [18]. The dehydrogenations of propylene on Fe1/MoS2 and Rh1/MoS2 via reaction 7 and reaction 8 are unfavorable because 1-propenyl and 2-propenyl are favorable for reverse hydrogenation to propylene due to the low barriers (Er ≈ 0.02~0.21 eV) (Table S4 in Supporting information) [33]. The reverse hydrogenation of 2-propenyl corresponds to the last elemental reaction mentioned in the non-reverse Horiuti-Polanyi mechanism reported by Xiao et al. [24]. Comparatively, Fe1/MoS2 exhibits better propylene selectivity than Rh1/MoS2 due to the low energy barrier for propylene desorption. The C–H bond cleavage on the terminal CH3 group is also considered for propylene dehydrogenation. As shown in Table S4 (Supporting information), the C–H bond scission on the CH3 group of propylene is quite feasible, indicating that deep dehydrogenation may initiate on any carbon atoms. Nevertheless, the activation energy to generate CH2=CHCH2 (Ef = 0.55 eV), which is an important intermediate state in deep dehydrogenation, is twice higher than that of the reverse reaction (Er = 0.17 eV). In addition, the dehydrogenation of CH2CHCH2 (Ef = 2.16 eV) is also much higher than the hydrogenation of CH2CHCH2 (Er = 0.17 eV) and propylene desorption of (1.48 eV), indicating that the propylene selectivity on Fe1/MoS2 can be assured even if deep dehydrogenation starts from the methyl group of propylene.
Based on the above screening calculations, Fe1/MoS2 is selected as a candidate catalyst for PDH. As shown in Fig. 2 and Table S5 (Supporting information), the activation barriers of propane and 1-propyl for C−C bond cracking are 1.65 eV and 1.80 eV, respectively, which are higher than the corresponding C−H bond cleavages (0.85 eV and 0.39 eV). Furthermore, the activation barrier of 1-propyl for deep dehydrogenation is 1.22 eV, which is higher than the corresponding C−H bond cleavage (0.39 eV). Therefore, the dehydrogenations of propane to propylene on Fe1/MoS2 can be hardly affected by side reactions.
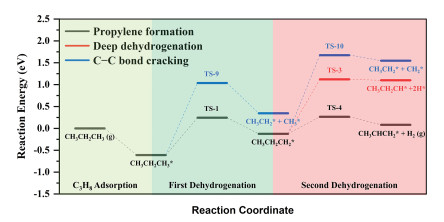
Notably, the PBE error originating from strong electronic correlations in transition metal based materials can be corrected with Hubbard-based DFT+U correction [34], hybrid functional (e.g., HSE06) [35] or many-body perturbation theory (GW) [36]. In this work, energy profiles of propane dehydrogenation on Fe1/MoS2 with DFT+U method [37] and RPBE functional [35] are calculated to study the effects of functionals. In DFT+U calculations, the Ueff value of 2.0 eV is applied for Fe 3d orbitals [38], while the U parameterization is not employed for the Mo 4d orbitals as suggested by previous theoretical studies [39-41]. As shown in Fig. S6 and Table S6 (Supporting information), activation barriers and reaction energies of the first C-H bond scission (Ef and ΔE of reaction 1) calculated with PBE+U are about 0.4 eV higher than those calculated with PBE. While other energies obtained with PBE, RPBE and PBE+U are quite close. Correspondingly, energy barriers for propane activations are significantly lower than those for deep dehydrogenation (Ef of reaction 7) and the reverse reaction of propylene dehydrogenation only need to overcome an ultra-low barrier of 0.14 eV in PBE+U calculations, indicating that the propylene selectivity can be assured with PBE, RPBE and PBE+U. Such computational results agree well with the conclusion of previous theoretical studies that the PBE+U correction is not exceedingly necessary for single-atom systems [42, 43].
Taking into consideration that the operating temperature of PDH is typically 770~980 K to achieve a reasonable propane conversion [24], the thermal stability of Fe1/MoS2 is evaluated by ab initio molecular dynamics (AIMD). As shown in Fig. S7 (Supporting information), no chemical bond is generated or broken for Fe1/MoS2 after 10-ps simulations, indicating that Fe1/MoS2 may maintain structural stability at temperatures as high as 800 K. Furthermore, we explore the Gibbs free energy profiles along the 1-propyl pathway on Fe1/MoS2 at 873 K, as shown in Fig. S8 and Table S5 (Supporting information). In DFT calculations, the temperature effect is included in Gibbs free energy G = E + ZPE – TS. PDH is an endothermic reaction in which both reaction energy and reaction entropy are positive. Therefore, high temperature is often employed in experiments to tune chemical equilibrium for a reasonable propane conversion efficiency. This trend can be seen clearly in our calculations. Kinetically, the collision of molecules will be accelerated. In the context of reaction barriers, the temperature effects are also shown in Table S7 (Supporting information). In conclusion, elevating temperatures will result in faster reaction kinetics and higher propane conversion efficiency. However, the propylene selectivity is not significantly affected.
Further analyses were conducted to reveal the catalytic and selective origins of Fe1/MoS2. First, the intrinsic electronic structure of Fe1/MoS2 is investigated. Fig. S9 (Supporting information) shows the spin-polarized density of states (DOS) projected onto d electrons of single Fe atoms on Fe1/MoS2, Fe atoms on the Fe (110) surface and an isolated Fe atom in vacuum. As expected, the localization of the d state of single Fe atoms on Fe1/MoS2 (red) is between the discrete atomic orbitals in an isolated Fe atom in vacuum (purple) and Fe atoms on the Fe (110) surface (green). The characteristic peaks in the energy window from −2.0 eV to 2.0 eV indicate that Fe1/MoS2 may exhibit higher catalytic activity than the Fe (110) surface.
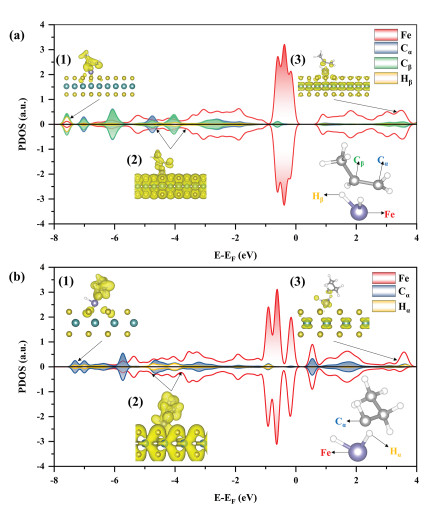
In addition to the energy profiles, the propylene selectivity can be clarified by bonding analysis of the transition states of reaction 3 and reaction 4 because the activation barriers and stability of the transition state (TS) are closely dependent on adsorbate-surface interactions [23]. Fig. 3 shows the PDOS of C atoms, H atoms and Fe active sites in the transition states of reaction 4 (TS-4) and reaction 3 (TS-3). As shown in Fig. 3a, the PDOS of the Hβ atom strongly overlaps with the Cβ atom of TS-4 in the energy windows of [−8.00, −7.00] eV and [−4.50, −3.50] eV, which are assigned as the bonding orbitals. The antibonding orbital is located in the energy window of [3.00, 4.00] eV. As illustrated in Fig. 3a(1), electrons accumulate not only between the Hβ atom and Cβ atom but also between the Hβ atom and disengaged H atom on Fe active sites, while electrons accumulate only between the Hβ atom and disengaged H atom in Fig. 3a(2), indicating that the Hβ atom of TS-4 chemically interacts with the Cβ atom and disengaged H atom. As shown in Fig. 3b, the PDOS of the Hα atom strongly overlaps with the Cα atom of TS-3 in the energy windows of [−7.50, −6.50] eV and [−5.00, −3.50] eV, which are often assigned as bonding orbitals. The antibonding orbital is located in the [3.00, 4.00] eV energy window. Different from TS-4, the electrons are only favorable for accumulation between the Hα atom and Cα atom, revealing that the Hα atom of TS-3 chemically interacts with the Cα atom, which explains the reverse process of deep dehydrogenation of 1-propyl.
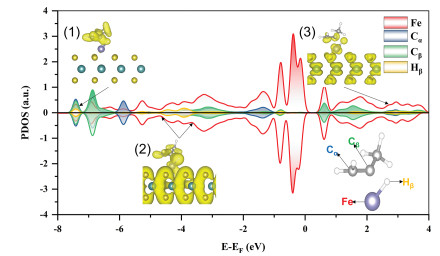
Inspired by the above bonding analyses, we further reveal the mechanism of the reverse hydrogenation of propenyl. As shown in Fig. 4, the PDOS of the Hβ atom strongly overlaps with the Cβ atom of TS-8 in the energy windows of [−8.00, −7.00] eV and [−5.00, −3.50] eV, which are often assigned as bonding orbitals. The antibonding orbital is located in the energy window of [2.50, 3.50] eV. Similar to TS-3, electrons always accumulate between the Hβ atom and Cβ atom, indicating that the Hβ atom only chemically interacts with the Cβ atom, which explains the reverse hydrogenation of propenyl. The reverse hydrogenation of propenyl inhibits dehydrogenations of propylene kinetically and ensures the fast desorption of propylene.
In summary, the PDH catalytic performances of TM1/MoS2 are investigated by DFT simulations. Fe1/MoS2 is selected as a candidate catalyst because the first C−H bond cleavage (rate-limiting step) can proceed with a low activation barrier being 0.85 eV, and the propylene selectivity can be assured by the kinetic inhibition of propylene dehydrogenation resulted from fast propenyl hydrogenation. PDOS and bonding analysis reveal that the Hβ atom of TS-8 is chemically interacted with the Cβ atom, which reveals the origin of the reverse hydrogenation of propylene. Calculations show that Fe1/MoS2 may exhibit excellent catalytic selectivity and can serve as a potential PDH catalyst. We believe that the unique catalytic selectivity of Fe1/MoS2 may inspire further investigations of on-purpose dehydrogenations of propane on SACs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank Natural Science Foundation of China (Nos. 21771134, 22173067), National Key R&D Program of China (Nos. 2017YFA0204800), Science and Technology Project of Jiangsu Province (No. BZ2020011), the Science and Technology Development Fund, Macau SAR (FDCT, No. 0052/2021/A), Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the 111 Project. X.F. thanks support from Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX20_2658).
Supplementary material associated with this article can be found, in the online version, at
J. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez, et al., Chem. Rev. 114 (2014) 10613–10653. doi: 10.1021/cr5002436
I. Amghizar, L.A. Vandewalle, K.M. Van Geem, et al., Engineering 3 (2017) 171–178. doi: 10.1016/J.ENG.2017.02.006
A. Agarwal, D. Sengupta, M. El-Halwagi, ACS Sustain. Chem. Eng. 6 (2018) 2407–2421. doi: 10.1021/acssuschemeng.7b03854
O. Onel, A.M. Niziolek, C.A. Floudas, Ind. Eng. Chem. Res. 55 (2016) 3043–3063. doi: 10.1021/acs.iecr.5b04571
S. Saerens, M.K. Sabbe, V.V. Galvita, et al., ACS Catal. 7 (2017) 7495–7508. doi: 10.1021/acscatal.7b01584
A. Al-Douri, D. Sengupta, M.M. El-Halwagi, J. Nat. Gas Sci. Eng. 45 (2017) 436–455. doi: 10.1016/j.jngse.2017.05.016
J.J. Siirola, AICHE J. 60 (2014) 810–819. doi: 10.1002/aic.14368
Z.P. Hu, D. Yang, Z. Wang, et al., Chin. J. Catal. 40 (2019) 1233–1254. doi: 10.1016/S1872-2067(19)63360-7
M. Szwarc, A.H. Sheon, J. Chem. Phys. 18 (1950) 237–238. doi: 10.1063/1.1747612
H. Fu, Z.P. Liu, Z.H. Li, et al., J. Am. Chem. Soc. 128 (2006) 11114–11123. doi: 10.1021/ja0611745
T. Shishido, K. Shimamura, K. Teramura, et al., Catal. Today 185 (2012) 151–156. doi: 10.1016/j.cattod.2011.10.028
J. Wu, Z.M. Peng, A.T. Bell, J. Catal. 311 (2014) 161–168. doi: 10.1016/j.jcat.2013.11.017
J. Im, M. Choi, ACS Catal. 6 (2016) 2819–2826. doi: 10.1021/acscatal.6b00329
Z.P. Han, S.R. Li, F. Jiang, et al., Nanoscale 6 (2014) 10000–10008. doi: 10.1039/C4NR02143F
H.N. Pham, J.J.H.B. Sattler, B.M. Weckhuysen, et al., ACS Catal. 6 (2016) 2257–2264. doi: 10.1021/acscatal.5b02917
N. Kaylor, R.J. Davis, J. Catal. 367 (2018) 181–193. doi: 10.1016/j.jcat.2018.09.006
X.Y. Sun, M.J. Liu, Y.Y. Huang, et al., Chin. J. Catal. 40 (2019) 819–825. doi: 10.1016/S1872-2067(18)63196-1
N. Kong, X. Fan, F. Liu, et al., ACS Nano 14 (2020) 5772–5779. doi: 10.1021/acsnano.0c00659
K.W. Sun, A.X. Chen, M.Z. Liu, et al., J. Am. Chem. Soc. 140 (2018) 4820–4825. doi: 10.1021/jacs.7b09097
J.J. Zhang, C.R. Chang, B. Yang, et al., Chem. Eur. J. 23 (2017) 6185–6189. doi: 10.1002/chem.201605744
H.M. Zhang, L.F. Chi, Adv. Mater. 28 (2016) 10492–10498. doi: 10.1002/adma.201602131
D.Y. Zhong, J.H. Franke, S.K. Podiyanachari, et al., Science 334 (2011) 213–216. doi: 10.1126/science.1211836
K.F. Niu, Z.H. Cj, Y.Y. Li, et al., J. Phys. Chem. C 123 (2019) 4969–4976. doi: 10.1021/acs.jpcc.8b12274
L. Xiao, Y.L. Shan, Z.J. Sui, et al., ACS Catal. 10 (2020) 14887–14902. doi: 10.1021/acscatal.0c03381
X.R. Cao, Y.F. Ji, Y. Luo, J. Phys. Chem. C 119 (2015) 1016–1023. doi: 10.1021/jp508625b
J. Li, S. Chen, F.J. Quan, et al., Chem. 6 (2020) 885–901. doi: 10.3390/polym12040885
T.H.M. Lau, X.W. Lu, J. Kulhavy, et al., Chem. Sci. 9 (2018) 4769–4776. doi: 10.1039/C8SC01114A
T. Bligaard, J.K. Norskov, S. Dahl, et al., J. Catal. 224 (2004) 206–217. doi: 10.1016/j.jcat.2004.02.034
J. Cheng, P. Hu, P. Ellis, et al., J. Phys. Chem. C 112 (2008) 6082–6086. doi: 10.1021/jp711051e
M.L. Yang, Y.A. Zhu, C. Fan, et al., Phys. Chem. Chem. Phys. 13 (2011) 3257–3267. doi: 10.1039/c0cp00341g
M.L. Yang, C. Fan, Y.A. Zhu, et al., J. Phys. Chem. C 119 (2015) 21386–21394. doi: 10.1021/acs.jpcc.5b03733
M.L. Yang, Y.A. Zhu, X.G. Zhou, et al., ACS Catal. 2 (2012) 1247–1258. doi: 10.1021/cs300031d
J.H. Yun, R.F. Lobo, J. Phys. Chem. C 118 (2014) 27292–27300. doi: 10.1021/jp504453n
S.L. Dudarev, G.A. Botton, S.Y. Savrasov, et al., Phys. Rev. B 57 (1998) 1505–1509. doi: 10.1103/PhysRevB.57.1505
S. Vijay, J.A. Gauthier, H.H. Heenen, et al., ACS Catal. 10 (2020) 7826–7835. doi: 10.1021/acscatal.0c01375
F. Aryasetiawan, O. Gunnarsson, Rep. Prog. Phys. 61 (1998) 237–312. doi: 10.1088/0034-4885/61/3/002
X. Ao, W. Zhang, Z.S. Li, et al., ACS Nano 13 (2019) 11853–11862. doi: 10.1021/acsnano.9b05913
S. Vijay, W. Ju, S. Bruckner, et al., Nat. Catal. 4 (2021) 1024–1031. doi: 10.1038/s41929-021-00705-y
T. Chanier, M. Sargolzaei, I. Opahle, et al., Phys. Rev. B 73 (2006) 134418. doi: 10.1103/PhysRevB.73.134418
R. Mishra, W. Zhou, S.J. Pennycook, et al., Phys. Rev. B 88 (2013) 144409. doi: 10.1103/PhysRevB.88.144409
A.N. Andriotis, M. Menon, Phys. Rev. B 90 (2014) 125304. doi: 10.1103/PhysRevB.90.125304
J.C. Liu, H. Xiao, J. Li, J. Am. Chem. Soc. 142 (2020) 3375–3383. doi: 10.1021/jacs.9b06808
F.Y. Li, Y.F. Li, X.C. Zeng, et al., ACS Catal. 5 (2015) 544–552. doi: 10.1021/cs501790v

Figure 1 The reaction energies of the first dehydrogenations of propane on TM1/MoS2. The dashed horizontal line (potential energy = 1.22 eV) is selected to indicate the corresponding value of the Pt-Sn catalyst in ref. [32].

Figure 3 (a) PDOS of the transition state in reaction 4 (TS-4). The insets show the partial charge densities in the energy ranges of (1) [−8.00, −7.00], (2) [−4.50, −3.50] and (3) [3.00, 4.00] eV. The values of the isosurface are 0.10 e/Å3. (b) PDOS of the transition state in reaction 3 (TS-3). The insets show the partial charge densities in the energy ranges of (1) [−7.50, −6.50], (2) [−5.50, −3.50], and (3) [3.00, 4.00] eV. The values of the isosurface are 0.10 e/Å3.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

