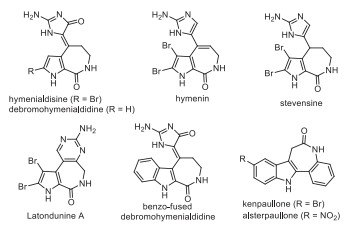
Figure 1.
Bioactive pyrroloazepinone derivatives.
Copper-catalyzed chemoselective heterocyclization of two isocyanides: Facile access to pyrroloazepinone derivatives
Xiaoyu Guo , Jinhuan Dong , Yunjie Zhu , Lan Bao , Zhongyan Hu , Xianxiu Xu

Pyrroloazepinone and its benzo-fused skeletons are found in a variety of bioactive natural products and synthesized compounds [1-6]. For example (Fig. 1), hymenialdisine is an inhibitor of cyclin-dependent kinase (CDK1 and CDK2) and glycogen synthase kinase-3b (GSK-3b) [1, 2]. Debromohymenialdisine is a good inhibitor of the cell cycle regulator checkpoint kinase 2 (CHK2) [3], and hymenin displays potent α-adrenoceptor blocking properties [4]. Additionally, benzo-fused debromohymenialdisine [5] and kenpaullone derivatives [6] exhibit comparable activities to their parent natural products. While considerable efforts have been devoted to their syntheses [7-11], new efficient protocols for the construction of pyrroloazepinones are still in high demand.
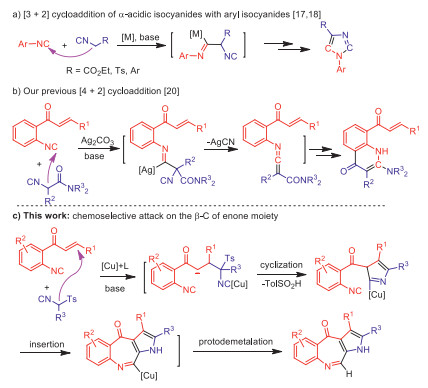
During the past decade, dimerization of two isocyanides has proven to be an efficient and useful tool for the construction of nitrogen-containing heterocycles [12-21]. In this context, the cycloaddition of α-acidic isocyanides with another aryl isocyanides has drawn considerable attention (Scheme 1) [16-21]. In 1999, Grigg and co-workers firstly disclosed the homo-cycloaddition of isocyanoacetate to produce imidazoles [16]. Six years later, an elegant cross-cycloaddition of aryl isocyanides with α-acidic isocyanides was achieved by the group of Yamamoto for catalytic synthesis of imidazoles (Scheme 1a) [17]. After that, Hong's [18] and Bi's [19] groups independently extended this reaction for preparation of 1, 4-diaryl- and 1, 4, 5-trisubstituted imidazoles (Scheme 1a). Recently, we found a [4 + 2] rather than [3 + 2] cycloaddition with α-substituted isocyanoacetamides and aryl isocyanides to access fused pyridones (Scheme 1b) [20]. Mechanistically, the above isocyanide-based [3 + 2] and [4 + 2] cycloadditions are all initiated by the nucleophilic attack of α-carbanion to the other isocyano group. Thus, the new reactivity profiles of these two isocyanides are still needed to explore for the divergent heterocycle syntheses. In continuation of our studies on isonitrile chemistry [22-25], we herein report a copper-catalyzed chemoselective heterocycloaddition of two different isocyanides for the synthesis of benzopyrroloazepinones (Scheme 1c). Unlike the previous isocyanide-based [3 + 2] and [4 + 2] reactions [20], this domino transformation is triggered by the chemoselective attack of substituted tosylmethyl isocyanides (sTosMIC) to the C=C double bond of o-cinnamoyl arylisocyanides. Notably, the azepinone ring is formed by an isocyanide insertion into the C–Cu bond of the in situ generated transient pyrrolocuprate intermediate.
Initially, o-cinnamoyl phenylisocyanide 1a and sTosMIC 2a was chosen as the model substrates to screen the reaction conditions (Table 1, for more details, see Supporting information). When 1a (0.1 mmol) and 2a (1.5 equiv.) was treated with CuI (5 mol%), dppe (7.5 mol%), and Cs2CO3 (1.2 equiv.) at 80 ℃ in THF for 10 min, benzopyrroloazepinone 3a (CCDC: 2124660) was obtained in 69% yield and the van Leusen-type pyrrole [26, 27] 4a in 15% yield (Table 1, entry 1). Only 80% yield of 4a was obtained without CuI (Table 1, entry 2), and the yield of 3a decreased to 41% without 1, 2-bis(diphenylphosphino)ethane (dppe) (Table 1, entry 2). Then, we screened the copper catalyst and found that other copper catalyst such as CuBr, CuOAc, and Cu2O gave a lower yield of 3a (Table 1, entries 4–6). It was showed that Cs2CO3 was more effective than DBU and t-BuOK (Table 1, entry 1 vs. entries 7 and 8). Solvent screening revealed that THF was the best choice (Table 1, entries 1 and 9–11). When the temperature was dropped to r.t., the yield of 4a was decreased obviously (Table 1, entry 12).
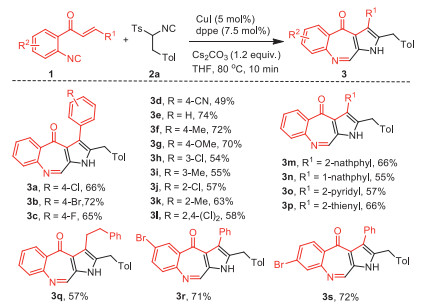
With the optimal conditions in hand, we examined the scope of isocyanides 1. When substrates 1 bear various R1 and R2 groups, all the tested reactions were completed within 10 min, providing the corresponding products 3a-3s in moderate to good yields (Scheme 2). Electron-poor, -neutral and -rich aryl groups afforded the products 3a-3c and 3e-3g in good yields (65%−74%). In contrast, substrates 1 with strong electron-withdrawing group substituted phenyl (1d, R1=4-CNC6H4) gave the fused azepinone 3d in lower yield (49%). Compared with the para-substituted aryl groups (1a-1c and 1e-1g), meta- (1h and 1i) and ortho-substituted aryls (1j and 1k) led to slight lower yields of azepinone 3h-3k. R1 group on isocyanides 1 also tolerated disubstituted aryl (1l), 1- and 2-naphthyls (1m and 1n), heteroaryls (1o and 1p) and alkyl groups (1q). Moreover, isocyanides 1r and 1s bearing bromo (R2) group at the different positions of phenyl also gave the corresponding products 3r and 3s in good yields, which may be served as handles for further diversification.
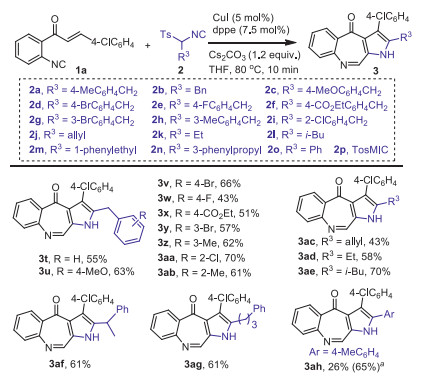
Subsequently, the scope of sTosMIC 2 was screened and the results were showed in Scheme 3. Generally, the heterocyclization tolerated a series of isocyanides 2 to generate a wide range of polysubstituted benzopyrroloazepinones 3t–3z and 3aa–3ah in moderate to good yields. sTosMIC 2 with various substituted benzyl and alkyl R3 groups, such as electron-neutral (2b), electron-rich (2c and 2 h), and electron-poor benzyl groups (2d‒2g and 2i), allyl (2j), ethyl (2k), i–butyl (2l), 1-phenylethyl (2 m) and 3-phenylpropyl (2n) groups were effective α-acidic isocyanide components and enabled the formation of fused azepinones 3t–3z and 3aa–3ag in moderate to good yields. When isocyanide 2o bearing a p-tolyl R3 group was employed, the corresponding benzopyrroloazepinone 3ah was obtained in 26% yield, along with the classic van Leusen-type pyrrole [26, 27] product 4ah in 65% yield. When TosMIC 2p was used as a substrate, only pyrrole 4ai was obtained in 77% yield.
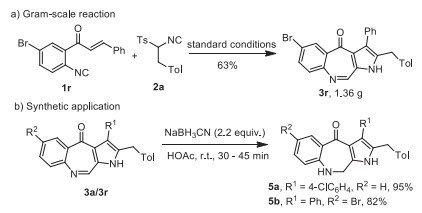
To evaluate the synthetic applicability of this transformation, a gram-scale synthesis was carried out. Under standard conditions, 1.36 g of product 3r was obtained in 63% yield from isocyanides 1r and 2a (Scheme 4a). On the other hand, the C=N in the pyrroloazazepines can easily be reduced by NaBH3CN (Scheme 4b). Dihydroazepinone derivatives 5a and 5b were obtained in 95% and 82% yields, respectively.
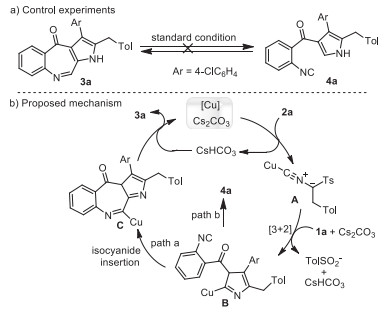
To figure out the mechanism, control experiments were executed (Scheme 5a). Under the standard conditions, pyrrole 4a and fused azepinone 3a could not convert to each other. This result demonstrates that the seven-membered azepinone ring is stable and the pyrrole 4a is not an intermediate for the formation of azepinone 3a. Based on the above experimental results and preceding studies [28-32], we proposed a conceivable mechanism (Scheme 5b, exemplified by the formation of 3a). First, coordination of CuI to isocyanide 2a, followed by the abstraction of a proton, forms a dipolar copper complex A [17, 28]. A formal [3 + 2] cycloaddition of intermediate A with the C=C double of isocyanide 1a occurs to generate the pyrrolocuprate intermediate B [29]. Then, isocyanide insertion into the C–Cu bond (path a) [28, 30-32] produces organocuprate C with construction of the seven-membered azepinone ring. Finally, the sequentially protodemetalation and aromatization result the final product 3a. The byproduct 4a is produced by protodemetalation of pyrrolocuprate intermediate B (path b).
In conclusion, we have developed a tandem chemoselective heterocyclization of two different isocyanides for the efficient synthesis of benzopyrroloazepinones. A tandem [3 + 2] cycloaddition and isocyanide insertion into the C–Cu bond of transient organocuprate intermediate process is proposed as the possible mechanism. The high efficiency of this methodology is exhibited by the formation of two rings and three bonds in a single step and the using of cheap metal catalyst. Further research is underway in our research group.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from the National Natural Science Foundation of China (Nos. 22001151, 22171168, 22101159), Taishan Scholar Program of Shandong Province and Shandong Provincial Natural Science Foundation (Nos. ZR2019QB004, ZR2020QB019) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Wan, W. Hur, C.Y. Cho, et al., Chemi. Biol. 11 (2004) 247–259.
C. Lange, E. Mix, J. Frahm, et al., Neurosci. Lett. 488 (2011) 36–40.
V. Sharma, J.J. Tepe, Bioorg. Med. Chem. Lett. 14 (2004) 4319–4321.
J. Kobayashi, H. Nakamura, Y. Ohizumi, Experientia 47 (1988) 86–87.
V. Sharma, T.A. Lansdell, G. Jin, J.J. Tepe, J. Med. Chem. 47 (2004) 3700–3703.
C. Schultz, A. Link, M. Leost, et al., J. Med. Chem. 42 (1999) 2909–2919.
H. Annoura, T. Tatsuoka, Tetrahedron Lett. 36 (1995) 413–416.
G. Papeo, H. Posteri, D. Borghi, M. Varasi, Org. Lett. 7 (2005) 5641–5644.
R.P. Singh, M.R. Bhandari, F.M. Torres, et al., Org. Lett. 22 (2020) 3412–3417.
F.X. Domínguez-Villa, G. Ávila-Zárraga, C. Armenta-Salinas, Tetrahedron Lett. 61 (2020) 151751.
S. Gracia, J. Schulz, S. Pellet-Rostaing, M. Lemaire, Synlett 12 (2008) 1852–1856.
M. Marin-Luna, M. Alajarin, Eur. J. Org. Chem. 34 (2020) 5496–5500.
Z. Hu, H. Yuan, Y. Men, et al., Angew. Chem. Int. Ed. 55 (2016) 7077–7080.
S. Su, J. Hu, Y.M. Cui, et al., Chem. Commun. 55 (2019) 12243–12246.
Z. Wang, X. Meng, P. Liu, W.Y. Hu, Y. Zhao, Org. Chem. Front. 7 (2020) 126–130.
R. Grigg, M.I. Lansdell, M. Thornton-Pett, Tetrahedron Lett. 55 (1999) 2025–2044.
C. Kanazawa, S. Kamijo, Y. Yamamoto, J. Am. Chem. Soc. 128 (2006) 10662–10663.
B. Pooi, J. Lee, K. Choi, H. Hirao, S.H. Hong, J. Org. Chem. 79 (2014) 9231–9245.
H. Wang, R.K. Kumar, Y. Yu, et al., Chem. Asian J. 11 (2016) 2841–2845.
Z. Hu, J. Dong, Y. Men, et al., Angew. Chem. Int. Ed. 56 (2017) 1805–1809.
Y. Gao, Z. Hu, J. Dong, J. Liu, X. Xu, Org. Lett. 19 (2017) 5292–5295.
X. Xu, L. Zhang, X. Liu, L. Pan, Q. Liu, Angew. Chem. Int. Ed. 52 (2013) 9271–9274.
L. Zhang, X. Xu, J. Tan, et al., Chem. Commun. 46 (2010) 3357–3359.
Y. Men, Z. Hu, J. Dong, X. Xu, B. Tang, Org. Lett. 20 (2018) 5348–5352.
J. Dong, L. Bao, Z. Hu, et al., Org. Lett. 20 (2018) 1244–1247.
L. Zhang, X. Xu, Q.R. Shao, L. Pan, Q. Liu, Org. Biomol. Chem. 11 (2013) 7393–7399.
L. Zhang, X. Xu, W. Xia, Q. Liu, Adv. Synth. Catal. 353 (2011) 2619–2623.
J. Du, X. Xu, Y. Li, L. Pan, Q. Liu, Org. Lett. 16 (2014) 4004–4007.
Q. Cai, F. Zhou, T. Xu, L. Fu, K. Ding, Org. Lett. 13 (2011) 340–343.
G. Qiu, Q. Ding, J. Wu, Chem. Soc. Rev. 42 (2013) 5257–5269.
D. Li, T. Mao, J. Huang, Q. Zhu, Chem. Commun. 53 (2017) 1305–1308.
J.W. Collet, T.R. Roose, E. Ruijter, B.U.W. Maes, R.V.A. Orru, Angew. Chem. Int. Ed. 59 (2020) 540–558.

Scheme 2 Scope of o-cinnamoyl arylisocyanide 1. Reaction conditions: 1a (0.2 mmol), 2a (1.5 equiv.), CuI (5 mol%), dppe (7.5 mol%), Cs2CO3 (1.2 equiv.), THF (2 mL), 10 min. Isolated yields of 3, and the reactions gave the corresponding pyrroles 4 in 10%−20% yields.

Scheme 3 Scope the α-acidic isocyanides 2. Reaction conditions: 1a (0.2 mmol), 2 (1.5 equiv.), CuI (5 mol%), dppe (7.5 mol%), Cs2CO3 (1.2 equiv.), THF (2 mL), 10 min. Isolated yields of 3, and the reactions gave the corresponding pyrroles 4 in 10%–20% yields. a Yield of pyrrole 4ah.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:


