引用本文:
肖洁琼, 吴小宁, 王倩, 王永妮, 贺怀儒. 过渡金属磷化物的制备及其催化性能研究进展[J]. 化学通报,
2021, 84(3): 215-224.

Citation: Jieqiong Xiao, Xiaoning Wu, Qian Wang, Yongni Wang, Huairu He. Progress in the Preparation and Catalytic Performance of Transition Metal Phosphides[J]. Chemistry, 2021, 84(3): 215-224.
Citation: Jieqiong Xiao, Xiaoning Wu, Qian Wang, Yongni Wang, Huairu He. Progress in the Preparation and Catalytic Performance of Transition Metal Phosphides[J]. Chemistry, 2021, 84(3): 215-224.
过渡金属磷化物的制备及其催化性能研究进展
English
Progress in the Preparation and Catalytic Performance of Transition Metal Phosphides
Abstract:
Recently, cheap and abundant phosphides have gradually attracted people's attention. Transition metal phosphides (TMPs) are very attractive due to their unique metal-like physical and chemical properties, high conductivity and good catalytic performance. It is widely used in metallurgy, hydroprocessing, electrocatalysis, energy storage, photocatalysis and other fields, becoming one of the hot points in the field of catalytic materials. This article mainly reviews the structural characteristics and commonly used preparation methods of TMPs and the latest developments in their applications in hydrorefining, electrocatalysis and photocatalysis.
-
Key words:
- Transition metal phosphide
- / Catalyst
- / Photocatalysis
- / Electrocatalysis
- / Hydrorefining
-
目前,催化反应常用的催化剂主要包括金属氧化物[1]、金属硫化物[2]、以及贵金属催化剂[3]等。但由于资源稀少、成本高昂或者化学性质并不稳定、抗毒性较低等原因,寻找廉价、丰富的非贵金属材料替代贵金属催化剂已迫在眉睫。1998年,Li等制备了一种具有良好加氢脱氮性质的催化剂MoP,开辟了一类全新的催化材料[4]。近年来,过渡金属磷化物(TMPs)作为一种新型催化材料,开始广泛应用在电化学[5]、生物学[6]、光学[7]、催化[8]等方面。因为其丰度高、成本低,存在丰富的活性位点,良好的导电性,结构稳定性和热稳定性[9],所以具有降低催化剂成本的巨大潜力。相比传统硫化物催化剂,TMPs拥有更加优异的加氢精制活性,在许多涉氢反应中表现出良好的催化性能[10~12]。此外,其在光催化和电催化等方面也展现出优异的性能,磷原子的插入不仅有利于光生电荷的捕获,而且可以有效地促进光生电荷的分离和转移[13]。在酸性或碱性介质中TMPs也对电催化裂解水的析氧反应(OER)和析氢反应(HER)表现出良好的催化活性。因此,开展对TMPs催化剂的研究具有重要的理论意义和潜在的应用前景,已经成为催化材料领域研究的新热点之一。
1. 过渡金属磷化物的结构特征
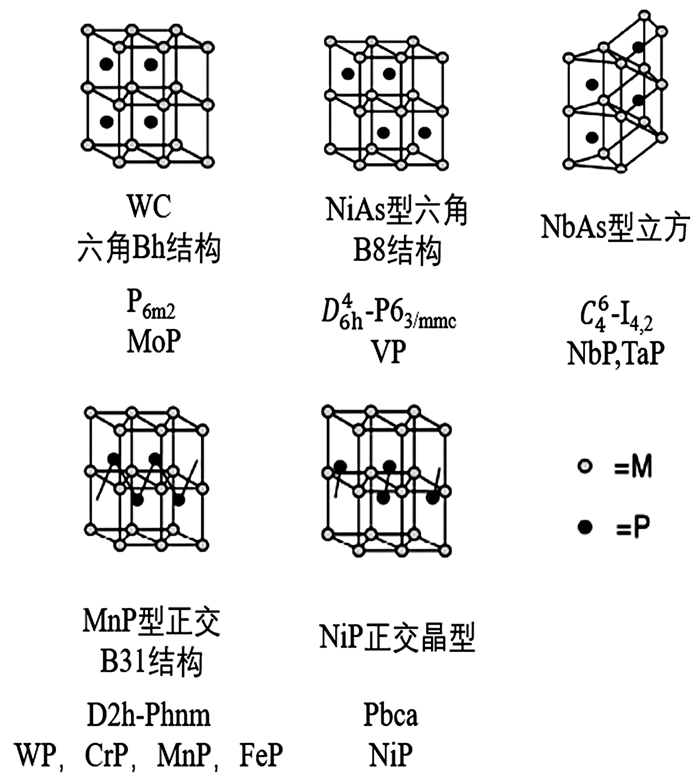
TMPs是通过磷原子进入过渡金属的晶格中而形成的一类具有特殊物理化学性质的间充化合物。磷化物的键合范围从与碱金属、碱土金属形成离子键化合物到与过渡金属形成金属键或共价键化合物,还可以与主族元素形成共价键化合物[14~17]。TMPs的理化性质与氮化物和碳化物基本相似,但是在晶体结构方面却有所不同,因此具有更优越的催化活性。磷化物中的磷原子半径(0.109nm)远远大于碳(0.071nm)或氮(0.065nm)的原子半径,使得磷原子不适合由封闭的金属原子形成普通八面体空穴,不能像碳化物和氮化物一样形成面心立方、密排六边形或简单六边形等相对简单的晶格[18]。因此,在金属磷化物中金属原子围绕着非金属原子形成三角形棱柱,非金属原子通常位于三角棱柱的中心。
图 1总结了不同的晶体形式[17]。MoP与WC呈等结构,非金属棱柱相互叠置。单磷化物VP具有Ni-As结构,V作简单六方棱柱体排列,P交替地填入一半的V的三方棱柱空隙,P棱镜横向位移为晶格间距的一半。单磷化物NbP和TaP也呈现紧密相关的Nb-As结构,但与VP的棱镜堆积方式不同。6~10族单磷酸盐采用MnP和NiP结构,这两种结构都可以看作Ni-As结构的畸变[18],MnP结构中磷原子形成链,而在NiP结构中磷原子形成对。TMPs在形貌和晶体结构上的差异都会对其性能产生显著影响。
图 1
2. 过渡金属磷化物制备方法
TMPs的制备方法有很多[19~21],已有诸多研究报道了合成磷化物的方法(表 1)[17]:(1)红磷单质与金属的直接化合反应[22];(2)金属卤化物与Na3P的固态置换反应;(3)金属卤化物与PH3的反应[23];(4)有机金属化合物的分解反应[24];(5)熔融盐的电解反应;(6)金属磷酸盐的还原[25]。然而在催化应用中,许多制备方法都不够经济,需要使用昂贵的反应原料,并且在高温高压等较苛刻的反应条件下进行,还可能会产生可能污染产品的副产物,如PH3,或者是方法不适用于制备负载型催化剂。下面将详细介绍几种制备TMPs的方法。
表 1
 下载:
导出CSV
下载:
导出CSV
方法 反应方程式 单质直接化合法 M0+ xP0(red)→MPx 固态置换反应法 MClx+Na3P→MP+NaCl 金属卤化物与PH3反应 MClx+PH3→MP+HCl+H2 有机金属化合物的分解 TiCl4(PH2C6H11)2→TiP+PH3+HCl+C6H10 熔融盐电解法 MOx+NaPO y →MP+Na2O 磷酸盐还原法 MPOx+H2→MP+ xH2O 2.1 溶剂热法制备TMPs
溶剂热法是在水热法的基础上逐步发展起来的,其利用有机溶剂作为反应介质,能够制备出具有特殊性质和结构的新型纳米材料[26]。Ranganatha等[27]对简单的一步溶剂热法进行了改进,合成了介孔NiCoP。这种具有孔洞和隧道的材料拥有巨大的比表面积,可以减少电荷/离子扩散路径,提高输运率,表现出优越的比电容和优异的循环性能。
Song等[28]开发了一种以三苯基膦(TPP)为磷源,以三正辛胺(TOA)为配位溶液相,在大气压下利用有序介孔材料MCM-41制备负载的Ni2P催化剂的新溶剂热方法,并研究了改变合成凝胶中的初始P/Ni摩尔比对其结构和加氢脱硫(HDS)性能的影响。结果表明,初始P/Ni摩尔比对磷化镍的结构和晶体尺寸影响很大,高的初始P/Ni摩尔比有利于形成小的且高度分散的Ni2P颗粒。此方法的还原温度相较于H2程序升温还原法(H2-TPR)低至少320K,并且所需原料比传统溶剂热法更为经济。
Wang等[29]首次开发了一种简便、温和的溶剂热方法,通过控制NaBH4和NaH2PO2的剂量选择性合成一系列NiPx,包括Ni、Ni12P5、Ni2P/Ni12P5、Ni/Ni2P和Ni2P,初步讨论了NiPx的形成机理。为获得最佳的NiPx助催化性能,进一步研究了制备的NiPx的化学计量对石墨氮化碳(g-C3N4)的光催化HER的影响。整个反应可以由式(1)简要描述。
$ 6 \mathrm{Ni}^{2+}+8 \mathrm{H}_{2} \mathrm{PO}_{2}^{-} \rightarrow 3 \mathrm{Ni}_{2} \mathrm{P}+\mathrm{P}+4 \mathrm{PO}_{4}^{3-}+16 \mathrm{H}^{+} $ (1) 结果表明,磷化过程主要是由NaBH4分解位置产生的活性[H]触发,将Ni2+依次还原为Ni0和将H2PO2-还原为P(在形成的Ni0附近),以及Ni0和原位生成的P原子之间的磷酸化。研究发现,NiPx的修饰可以显著提高g-C3N4的光催化活性且NiPx的助催化效率主要由P占比决定。Ni2P表现出最高的P含量,从而表现出最高的助催化性能。
在溶剂热法中通常采用低价态的含磷化合物作为制备TMPs的磷源,其具有还原性或可以发生歧化反应以生成具有还原性的物质[30]。该方法可通过对催化剂结构形貌等晶体特征的有效控制以提高催化性能,形成对反应更为有利的催化剂结构,且操作简单,反应周期较短,更为经济[31]。然而,由于在溶剂热法反应过程中可能会发生TMPs的团聚现象,使活性组分分散度降低。因此由水热、溶剂热法生成的催化剂产物一般无法满足工业化的要求。
2.2 热分解法制备TMPs
与传统的H2-TPR法相比,次磷酸盐热分解法[32]具有工艺简单、不需高温高压、原料和设备成本低等优点。
受Ni(CH3COO)2·4H2O在惰性气体中加热时,乙酸根热分解产生的CO会将Ni2+态还原为Ni的启发,Dai等[33]在不同温度下流动的N2气体环境中,以活性炭为载体,Ni(CH3COO)2·4H2O为Ni源,利用次磷酸铵(NH4H2PO2)的热分解制备了一系列Ni2P/C催化剂,研究了温度对催化剂结构表征和苯并呋喃(BF)催化加氢脱氧(HDO)活性的影响。结果表明,随着制备温度的升高,更多的活性Ni位点暴露在催化剂表面,磷的富集减少,Ni2P/C-x(x=制备温度,℃)催化剂的Ni2P晶粒尺寸和CO吸收量增加,Ni2P/C-x催化剂的BF转化率和总无氧产物的产率也随制备温度的升高而增加,且发现制备温度对反应路线的影响并不显著。
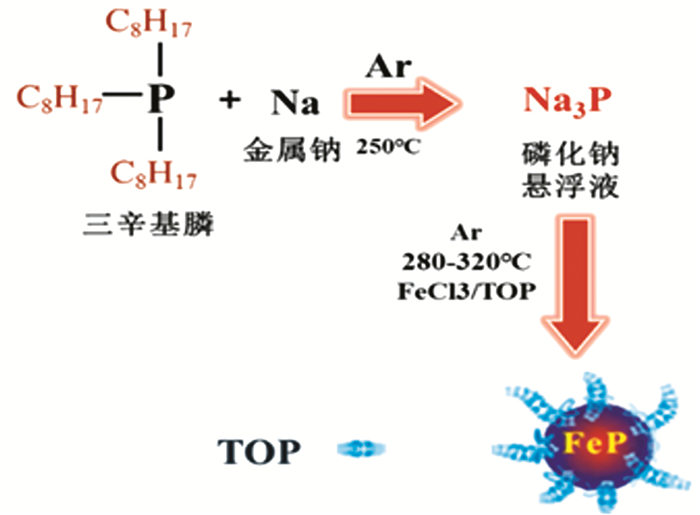
此外,三辛基膦(TOP)也是具有良好前景的磷源,已被用来合成多种金属磷化物。TOP热分解法[34]得到的物质颗粒小,粒子分散均匀,比表面积大,特别是能够得到具有特殊形貌和结构的材料如纳米空心球,因此也是研究的热点之一。Ahluwalia等[35]报道了一种新颖的一步热分解合成方法(图 2),使用TOP作为磷源和封端试剂来控制颗粒大小,生产结晶性TOP封端的磷化铁纳米颗粒。该方法提供了一种更为绿色的合成Na3P的替代途径,在钠存在下通过催化裂解P-C键在反应过程中产生游离磷,最后在250℃的氩气环境下与TOP反应形成Na3P。随后,合成的Na3P在TOP存在下进一步与FeCl3反应形成FeP/TOP-NPs。与同类产品相比,该产物FeP/TOP-NPs具有显著的超顺磁性,拥有磁性纳米粒子的典型特征。
图 2
通过热分解法制备TMPs,仅仅需要常压下简单的热处理,不需要高温、高压、程序升温等复杂的步骤,而且所用原料价格便宜、安全,所需设备成本低,过程中不会造成环境污染,适合大规模工业生产。
2.3 程序升温还原(TPR)法制备TMPs
TPR法制备TMPs是以高价态的磷酸盐或亚磷酸盐为前驱体,在氢气环境下将金属和磷在原子尺度上混合。Li等[4]在1998年首次采用TPR法制备了具有中等比表面积的单相MoP,发现其具有良好的加氢脱氮(HDN)催化性能。后续其他学者通过该方法制备了一系列磷化物,如Ni2P[36]、MoP[37]及双金属的CoNiP、CoMnP和CoCuP[38]。
TPR法制备TMPs的反应过程中,温度、原料配比、升温速率等因素都会对最终产物的组织形貌产生很大影响[39]。Pan等[40]通过TPR法在不同温度下煅烧磷酸盐前体制备了SiO2负载的Ni-Mo双金属磷化物。H2-TPR结果表明,当前驱体分别在400和500℃下煅烧时,制备的催化剂中除Ni2P和MoP相外,还会生成NiMoP2相。然而,当前驱体分别在600、700和800℃下煅烧时,制备的催化剂中只能检测到Ni2P和MoP相。研究发现,随着前驱体煅烧温度的升高,由于前驱体中Ni和Mo之间的相互作用减少以及金属磷酸盐颗粒的烧结,前驱体的还原变得困难,磷化物微晶尺寸趋于增大,随后导致表面金属位点密度和酸量降低。在月桂酸甲酯的脱氧中,前体在600℃下煅烧制备的催化剂表现出最佳性能。
由于TMPs的比表面积较小,所以通过负载能增加活性组分的分散度,使其在催化过程中的利用率提高[41]。TMPs的制备过程和催化性能都会受载体的表面性质影响。Lee等[42]通过TPR法制备了均匀分散在N掺杂的碳纳米管(CNT)和石墨烯复合材料(NCNT-NGR)载体上的10nm MoP纳米颗粒。NCNT-NGR载体由石墨烯层间插层良好的碳纳米管组成,抑制了CNT的成束和石墨烯的再堆积,在TPR过程中实现了碳载体的N掺杂,从而提供了较大的比表面积。在制备的催化剂中,MoP/NCNT-NGR催化剂在酸性介质中表现出最高的催化性能,在碱性介质中也观察到较高的HER活性。MoP/NCNTNGR催化剂的这种HER性能来自于高活性MoP纳米粒子与大表面积高导电NCNT-NGR载体之间的协同作用。
Zhang等[43]通过TPR法利用磷酸盐前驱体合成了Ni2P/SiO2-TPR催化剂样品,并以TOP为磷化剂,采用液相法(LP)制备了Ni2P/SiO2-LP样品。两种方法均得到Ni2P六方相的相纯材料,但TPR制备的样品的粒径(7nm)小于LP合成制备的样品(25nm)。随后在0.5MPa和250~450 ℃下,利用2-甲基呋喃(2-MF)在H2中进行了气相氢脱氧实验,结果发现Ni2P/SiO2-TPR比Ni2P/SiO2-LP具有更高的转化率和转换频率,并通过X射线光电子能谱测定了P/Ni比值,证实了二者活性的差异是由于TPR制备的样品磷含量较高所致。
TPR法制备TMPs不需要昂贵的化学试剂和较高的合成条件且更为绿色环保,所以一直受到科学研究者的青睐。
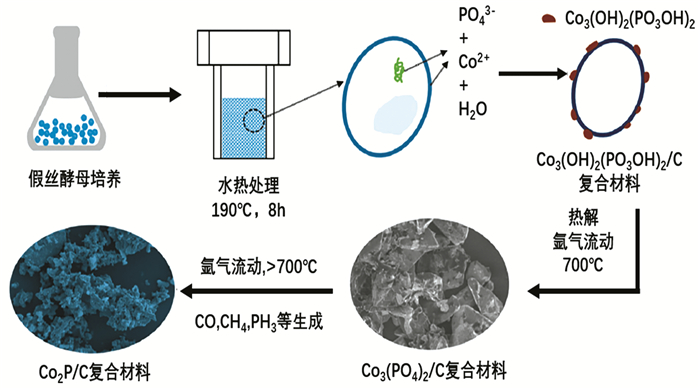
上述是几种制备TMPs的常用方法,近年来,科研人员也提出许多新的合成手段。Tong等[44]选择了一种易于培养且高生物量的假丝酵母作为模型微生物,利用发酵培养基的简单调节实现产菌中磷的积累;以收获的富磷微生物作为前驱体,通过水热处理和热解的联合方法形成磷化钴/碳复合材料(图 3)。Ma等[45]报道了一种利用强飞秒激光辐照硫酸镍和次磷酸钠水溶液制备非晶态磷化镍纳米粒子的方法。通过改变反应参数,如离子浓度、离子摩尔比、激光功率和辐照时间,调节纳米颗粒的形貌和尺寸,所合成的非晶态镍磷纳米粒子对高氯酸铵的热分解具有良好的催化性能。Zhang等[46]提出了一种简单有效的红磷自模板湿化学法制备层状纳米多孔金属磷化物的方法。通过改变离子液体(ILs)的加入量,控制阳离子的扩散速率和红磷模板剂的分解速率,制备出孔径和孔隙率可调的多孔磷化镍。合成的样品在高催化电流、低过电位下对OER具有显著的电催化活性以及良好的稳定性。
图 3

总之,TMPs的合成逐渐在向纳米化、高比表面化发展[47~49],如何增强材料的性能,开发具有改进特性和功能的廉价材料逐渐成为人们所关注的问题。一方面所利用的合成方法对TMPs的形成和结构中磷的分布起着至关重要的作用,可以通过控制颗粒尺寸、表面结构和颗粒分布,以制备具有改进特性的材料。另一方面,TMPs的表面积和孔径也会受不同合成条件的影响,例如前体材料的类型、磷化温度、加热速率和反应持续时间。还可以通过合成三元磷化物系统、掺入不同元素而改变主体材料的晶体结构或合成金属有机骨架(MOF)衍生的复合材料等使材料提供更大的表面积并暴露更多的催化活性位点,提高TMPs的催化活性。通过不同的途径合成会制备出不同结构的TMPs,制备时需要根据催化剂具体的应用对象、原料的性质以及合成设备等因素来选择合适的方法。
3. 过渡金属磷化物的催化性能
金属磷化物的结构以三角棱柱为基础,能很好地容纳较大的磷原子。与硫化物不同,磷化物不会形成层状结构,由于缺少层状结构所以使晶体形态的各向同性更加明显,因此有可能允许其更大程度地进入微晶表面上的活性位点和边缘部位。而且,表面金属原子能更好地暴露在液相反应物中[17],使其具有良好的催化性能。下面详细介绍TMPs在加氢精制、电催化以及光催化方面的应用。
3.1 加氢精制中的应用
随着工业的迅猛发展、原油的不断开采,伴随而来的是严重的工业污染和原油质量的急速下降。为了达到环保法规低硫、低氮的新标准,油品深度加氢精制成为唯一的解决途径。然而,传统的精制催化剂已经开始难以满足需要,对原催化剂进行改进或者开发新型催化剂的需求逐渐迫切。因此,TMPs作为一类新型加氢处理催化剂,在加氢脱硫、加氢脱氮和加氢脱氧等方面的催化应用得到了人们的广泛关注。
d’Aquino等[50]提出一种利用氢氧化镍(Ni(OH)2)和次磷酸(H3PO2)反应生成的次磷酸盐基前驱体在二氧化硅、氧化铝和无定形硅氧化铝(ASA)载体上原位制备Ni2P的方法。结果显示,Ni(H2PO2)2生成Ni2P的反应发生在468~561K,未负载的Ni(H2PO2)2在这个温度范围的低端发生反应,而铝负载的Ni(H2PO2)2在这个温度范围的高端反应。虽然不同方法制备的Ni2P/SiO2催化剂具有相似的HDN和HDS活性,但原位制备的Ni2P/Al2O3和Ni2P/ASA催化剂的活性明显高于由次磷酸酯和磷酸盐基前驱体原位制备的催化剂。
Galindo-Ortega等[25]以氯化钌和磷酸盐为前驱体,采用常规制备方法合成了负载在中孔二氧化硅(SBA-15)上的不同摩尔比(P/Ru)的活性磷化钌催化剂(样品标记为x-P/Ru,x为摩尔比值)。研究了不同摩尔比下对3-甲基噻吩(3MT)HDS活性的影响,其中3-P/Ru催化剂的HDS活性高于其他摩尔比催化剂,具有较高的异构化能力,表征发现是由于3-P/Ru催化剂酸度适中,表面上RuP和Ru2P活性相的分散度高且均匀,有助于3MT分子的吸附和H2的活化。
Rodríguez-Aguado等[11]合成了含铁量为15%的二氧化硅负载的磷化铁催化剂,研究了磷含量对铁基催化剂性能的影响,以及其在苯酚HDO反应中的活性。通过控制不同的磷含量得到了Fe2P、FeP和FeP2相。表征结果表明,在合成中初始磷/铁比不仅决定了磷化铁的化学计量,而且还决定了颗粒大小、金属表面暴露程度和酸度。具有唯一相的催化剂在苯酚的HDO反应中表现出较好的活性。此外,Fe2P相在HDO转化方面优于FeP相。
Song等[51]采用两种不同的磷源合成了MCM-41负载型高活性磷化镍HDS催化剂,催化剂表面由O2/N2混合物钝化改为空气改性,无需像传统方法在HDS反应前进行预处理。结果表明,空气改性可以促进催化剂表面形成更小的Ni2P颗粒和更活跃的Ni位点。与传统方法制备的催化剂相比,空气改性的催化剂具有更高的HDS活性。其原因在于较小的Ni2P颗粒尺寸和由于表面改性而增加的氢解离活性。
Inocencio等[52]制备了五种不同的无载体TMPs:Ni2P、MoP、CoP/CoP2、FeP和WP,并进行了气相中苯酚的HDO测试。所有催化剂均显示出对直接脱氧产物的高选择性,这表明金属磷化物相促进了C-O键的直接裂解。苯酚在金属磷化物颗粒上的强烈吸附降低了C-O键断裂的能垒,同时发现金属磷化物的类型显著影响产物分布。对于未负载的Ni2P催化剂,苯的选择性最高,而环己烯主要在CoP和WP上形成。在FeP上生成了环己烯以及环己烷,而在MoP上生成了C5-C6烃。因此,TMPs催化剂具有使苯酚脱氧以选择性产生芳族化合物的巨大潜力。
3.2 电催化反应中的应用
对化石燃料的持续消耗已导致其储量不断下降,能源需求日趋紧张,开发绿色、环保可再生的新能源迫在眉睫。氢是一种理想的新能源,电解水是获取氢能的重要方法之一。因为金属中心和磷位均具有高催化活性,近年来,TMPs被用作一种新型的电催化剂。由于其具有较高的电负性,可以从金属原子中吸取电子,所以TMPs中的磷原子含量对H2的生成起重要作用,其研究对于整体水分解而言是有吸引力且具有挑战性的。
Ding等[53]选择了三种不同组分的铁基普鲁士蓝类似物(PBA)作为前驱体,通过热处理调节磷化温度,得到了一系列FeCoP、FeNiP和FeMnP,其中400℃下制备的FeCoP具有最明显的多孔结构和最宽的孔径分布,有利于OER过程中的传质和放氧。此外,与Ni和Mn相比,Co的引入大大降低了TMPs的电荷转移电阻(Rct),加速了电子在OER中的输运。FeCoP-400(FeCoP-x,x=制备温度,℃)具有多孔的几何结构和独特的电子结构,在电流密度为10mA·cm-2时,具有261mV的过电位,在1mol/L KOH中表现出优异的、稳定的电催化活性。
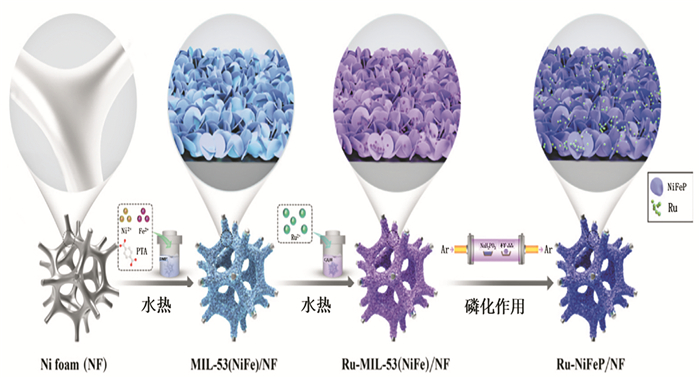
Lin等[54]开发了由镍泡沫上的二维MIL-53(NiFe)MOF纳米片衍生的超低钌(Ru)(0.6(wt)%)掺杂的双金属磷化物(即Ru-NiFeP/NF)(图 4), 其具有优化的电子结构和更强的电导率,在宽pH范围内具有优越的HER催化性能,酸性、中性和碱性介质中分别仅需要29、105和56 mV的过电势就能达到10mA·cm-2的电流密度。对于OER,仅需要179mV的过电势即可在碱性介质中达到10mA·cm-2的电流密度。这项工作强调了杂原子掺杂对MOF衍生物的合理设计和电子结构工程策略,可以扩展到设计和制备其他高性能的基于MOF的电催化剂。
图 4

分层的纳米结构由于其独特的性能而引起了人们的广泛关注,被认为是有前途的电催化材料。Hu等[55]使用自组装方法制备了新型的磷化钴纳米笼@铁锌混合金属磷化物纳米管的分层多孔纳米复合材料(CoP@ZnFeP)。由于其结构和组成优点,所制备的磷化物杂化物具有丰富的催化活性位点和高孔隙率,便于物质扩散。在碱性电解质中,与CoP和ZnFeP纳米粒子的机械混合物相比,CoP@ZnFeP花状杂化物显示出对OER和HER更强的催化活性。其优异的性能主要归因于纳米结构单元具有较大的表面积和轮廓分明的多孔通道,有利于电解质的传质和电荷传输。
Yang等[56]通过控制前体中仅有较高活性的Ni原子的磷化以构建Ni2P/MoO2/NF异质结构纳米棒阵列电催化剂(Ni2P/MoO2/NF HNRs),实现了选择性磷化策略。通过有效的界面构建,电子自发地跨Ni2P/MoO2杂交界面转移,产生强电子耦合效应,从而调节电子结构并增强固有催化活性,在碱性电解质中产生出色的电催化活性和耐久性。Ni2P/MoO2/NF HNRs在10mA·cm-2的析氢反应中表现出34mV的超低超电势。结果表明,强电子耦合作用增强了Ni2P和MoO2之间的协同作用,有利于提高H2的产生。
研究发现,磷酸盐对磷化物的电化学性能也具有有益的影响。He等[57]通过Ni-Co层状双氢氧化物(Ni-Co LDH)的磷化和磷酸盐的原位形成而制备了NiCoP磷酸盐纳米笼(NCPP NCs)催化剂。由于良好的表面润湿性和中空结构提供的充分暴露的活性中心,NCPP NCs对OER和HER均表现出优异的电催化活性以及良好的长期稳定性。在0.1mol/L KOH中,OER和HER分别仅需要291和140 mV的过电势,就可以实现10mA·cm-2的电流密度。作为双功能催化剂的NCPP NCs在整个水分解过程中表现出出色的性能,在10mA·cm-2下的电势仅为1.60V,并且具有长期稳定性。
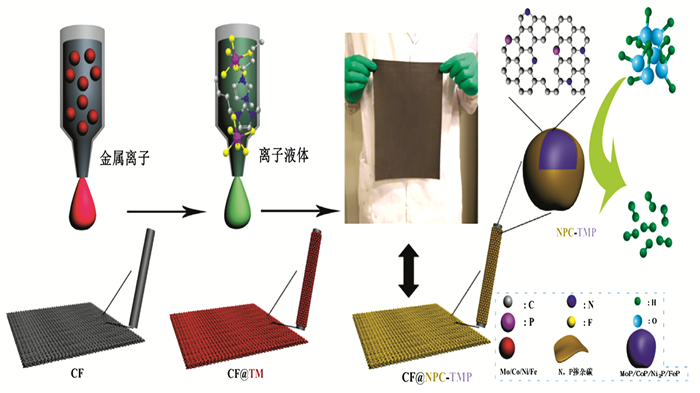
Xiao等[58]用一种简单的喷墨印刷技术合成了一系列封装在碳纤维布上的N、P共掺杂碳中的过渡金属磷化物纳米粒子(CF@NPC-TMP,TM=Mo,Co,Ni,Fe,图 5)作为高效氢析出阴极,所有制备的CF@NPC-TMP电极在很宽的pH范围(pH=0~14)内对HER均显示出理想的电催化性能和稳定性。CF@NPC-MoP电极仅需要在87和145 mV的过电势下就可以分别驱动0.5mol/L H2SO4中10和100 mA·cm-2的阴极电流密度。原位掺杂的碳纳米层不仅避免了TMP纳米粒子在高温下发生团聚,也提高了催化剂的电导率,以促进电荷快速转移到催化活性位,并且可防止TMP纳米粒子在HER期间腐蚀。
图 5
传统的催化剂在电池的大规模实际应用中仍占有不可动摇的地位。然而,昂贵而稀有的Pt、Li资源极大地限制了电极材料的应用,无法满足未来可持续能源转换和储存的迫切需求。因此,开发一些具有高活性、廉价耐用、电势低且资源富裕的催化材料势在必行。
3.3 光催化反应中的应用
光催化水分解制氢作为解决全球能源和环境问题的理想方法也受到了研究人员的广泛关注。TMPs由于其独特的结构在光催化制氢中可以起到增强光吸收、提供活性位点、加速电荷转移和增强光稳定性等作用。开发高活性、长期稳定、低成本的光催化剂仍是其商业应用的关键。
Tang等[59]将MoP纳米颗粒用作新型的助催化剂来制备g-C3N4负载的复合材料光催化剂,用于在模拟辐射下还原CO2和水分解。结果表明,该复合材料具有很高的活性,并表现出优异的稳定性。全面的特征分析表明MoP的引入促进了光生电子-空穴对的分离和转移,并且通过密度泛函理论(DFT)进行的理论计算也证实MoP可以有效地将光激发电荷与g-C3N4分离,且引入MoP纳米颗粒增加了活化的比表面积。该研究提供了一种设计具有成本效益的光催化剂的新方法。
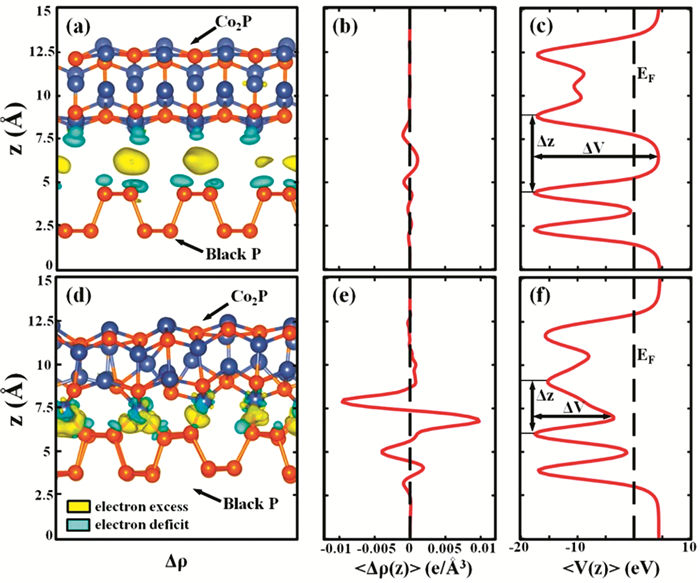
高的光诱导电荷-载流子分离效率在确定光催化剂的光催化活性方面起着至关重要的作用。Yuan等[60]通过将Co2P助催化剂生长到黑磷(BP)纳米片的边缘上,在Co2P/BP纳米片光催化剂中通过Co-P键来以精确的方式控制电荷分离(图 6)。Co-P键起到促进原子级电荷流控制的作用,可以改善BP纳米片与Co2P助催化剂之间的光生电荷-载流子转移,从而改善Co2P的光催化性能。Co2P/BP纳米片光催化剂的光催化H2生成速率是裸BP纳米片的39.7倍。此外,在BP纳米片边缘上生长的Co2P抑制了BP纳米片的降解,使得其对光催化产H2具有良好稳定性。
图 6

p-n结结构是开发有效的异质结构光催化剂的有效策略。Qin等[61]将p型磷化铜(Cu3P)与n型石墨氮化碳g-C3N4通过固相反应合成了一种新型且低成本的Cu3P/g-C3N4 p-n异质结光催化剂。形成的p-n结可以有效改善光诱导的电荷分离,以加速电荷转移并减少电荷复合,从而增强了光催化活性。优化的Cu3P/g-C3N4 p-n异质结光催化剂的活性是裸g-C3N4的95倍,在420nm处的表观量子效率为2.6%。开发有效的异质结结构光催化剂来加速电荷的分离和转移,对于改善利用太阳能的光催化氢的产生至关重要。
Man等[62]提出过渡金属铁、钴、锰和钼可以作为单相原子取代磷化镍的晶格中的镍,而不明显改变其金属结构和形态,制备了具有金属性质的过渡金属掺杂的磷化镍纳米颗粒。相比原始磷化镍纳米颗粒其在电催化HER和光催化方面都有改进,显示出高度可调的活性。氢覆盖率较低的钼掺杂磷化镍纳米粒子在电催化制氢反应中具有最佳的制氢性能,并且在有机光敏剂的作用下也表现出优异的光催化制氢能力。
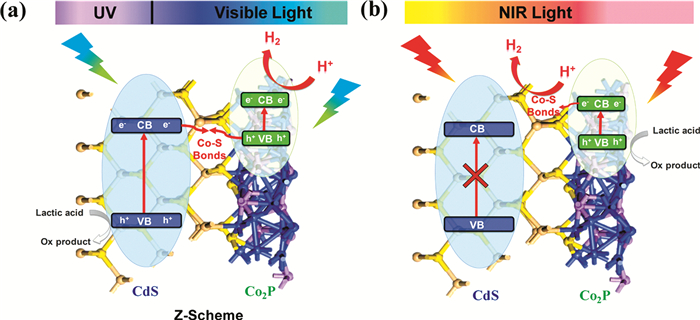
Li等[63]制备了不含贵金属的Co2P/CdS Z-scheme光催化剂,以实现超宽的紫外-可见-近红外光谱收集。与Pt/CdS、CdS和Co2P相比,优化的Co2P/CdS表现出更出色的性能,10%Co2P/CdS光催化剂就可以在太阳光、可见光和近红外光下有效分解水,HER最高速率分别高达262.16、66.98和3.93mmol·g-1·h-1。在700nm处10%Co2P/CdS的表观量子效率值可高达2.26%。Z-scheme转化途径可有效增强Co2P/CdS中用于紫外-可见光驱动的HER的载流子分离,并且研究发现界面处的Co-S键可以充当电荷转移的“桥梁”,从而增强全光谱驱动的H2产生(图 7)。
图 7

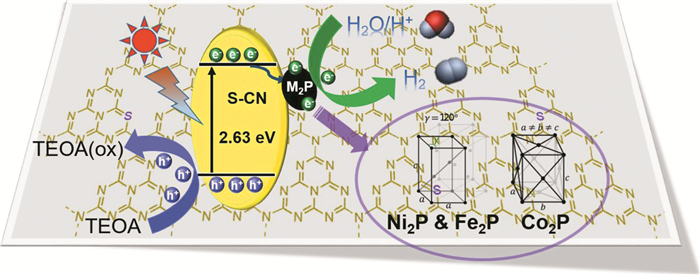
为了研究不同金属的磷化物对光催化生成H2的影响,Sun等[64]采用三种类型的铁基TMPs(M2P, M=Fe,Co,Ni)纳米颗粒作为硫掺杂g-C3N4(S-CN)光催化生成H2的助催化剂(图 8)。光致发光(PL)光谱、光电化学表征和DFT计算证实,有效的H2生成活性主要归因于从光诱导的S-CN到M2P的最低的过电势和有效的电荷转移,这抑制了S-CN中电子-空穴对的重组。当S-CN与Ni2P结合时,增强了光致电荷转移和带分离,从而具有最高的光催化H2生成活性。将二氧化碳光还原为碳氢化合物并将光分解水生产氢气是减少环境污染和太阳能转化为化学能的可持续的方法。然而,开发低成本、高效和稳定的光催化剂仍然是巨大的挑战。
图 8

染料是工业废水中最重要的污染物之一,利用半导体纳米材料的多相光催化活性降解废水中的有机污染物已经引起了广泛的兴趣[65]。Farahi等[66]采用一步水热法制备了具有不同结构相和形态的磷化镍纳米颗粒,结果表明,纳米结构的晶相和形态会影响样品的催化性能。低P/Ni摩尔比的Ni12P5相具有正方结构,高P/Ni比的Ni2P相具有六方结构。由于光催化过程发生在纳米材料的表面,具有Ni2P的结构相由于其多孔结构增加了有效面积,提高了磷化镍的光催化活性。P/Ni摩尔比为35/1且具有纯Ni2P相的样品去除亚甲蓝染料的效果最佳; 两相共存会降低光催化性能。
尽管有关研究已经发现TMPs具有降解废水中多种有机污染物并将其转化为简单的气态产物(H2O、CO2)和无机离子的高潜力,但通过TMPs催化降解废水中的环境污染物进行污水处理和水净化的有关研究还较少。TMPs作为新型催化剂仍具有很大的发展空间,其在水处理中的催化应用还有待进一步探索研究。
4. 总结和展望
尽管在涉及加氢精制、催化产氢等方面有关TMPs的研究已经取得了一些成就,但开发具有改进性能的TMPs材料仍然存在许多挑战。
(1) 应该开发更安全、更简单的合成方案,利用毒性较小且经济的材料来更加高效地合成TMPs。制备TMPs的大多数合成方法都需要较长的反应时间、较高的反应温度,同时,有毒的尾气和残留的有机溶剂会增加环境风险。因此迫切需要开发新的合成方案以安全、快速地制备TMPs,使其应用更加广泛。
(2) 应探索新型的TMPs催化剂和半导体。大多数有关TMPs的研究都基于镍和钴基材料,进一步的工作应包括研究基于其他过渡金属的磷化物;同时,除了将TMPs与常用的TiO2、CdS和g-C3N4等结合使用外,还应考虑其他新型半导体。
(3) 应尽量采用其他支撑材料掺杂或制备复合材料等合成方法,开发具有不同形态和结构的高活性TMPs。当前,用于催化产氢的大多数TMPs的形态是纳米粒子,易发生自聚集。因此需要开发具有丰富活性位点的纳米结构TMPs,提高催化效果。
随着经济的快速发展,当前的全球环境危机和能源危机已经成为全人类的重大问题,改善化石类能源的清洁性和寻求新的清洁能源已成为可持续发展的必由之路。因此对基于地球上储量丰富元素的性能优异的催化剂的探索具有重大意义。TMPs特殊的晶体结构和理化性质使其具有良好的催化性能,在加氢精制、电化学催化制氢、光催化等不同领域的应用得到广泛的研究。通过研究表明,开发适用于各种催化反应的高效、廉价、稳定的TMPs是十分可行的。相信随着新型合成策略的发展和机理的阐明,TMPs在催化反应中的商业意义也将大大提高。
-
-
[1]
Zhao X, He X, Chen B, et al. Appl. Surf. Sci., 2019, 487: 1049~1057. doi: 10.1016/j.apsusc.2019.05.182
-
[2]
Diao X, Ji N, Zheng M, et al. J. Energy Chem., 2018, 27(2): 600~610. doi: 10.1016/j.jechem.2017.07.008
-
[3]
Messou D, Vivier L, Especel C, et al. Fuel Proc. Technol., 2018, 177: 159~169. doi: 10.1016/j.fuproc.2018.04.030
-
[4]
Li W, Dhandapani B, Oyama S T. Chem. Lett., 1998: 207~208.
-
[5]
Maduraiveeran G, Sasidharan M, Jin W. Prog. Mater. Sci., 2019, 106: 100574. doi: 10.1016/j.pmatsci.2019.100574
-
[6]
Luong D, Yergeshov A A, Zoughaib M, et al. Mater. Sci. Eng. C, 2019, 103: 109759. doi: 10.1016/j.msec.2019.109759
-
[7]
Wen H, Xie S, Cui J, et al. J. Lumin., 2019, 213: 263~272. doi: 10.1016/j.jlumin.2019.05.016
-
[8]
Massel F, Ahmadi S, Hahlin M, et al. J. Electron. Spectrosc., 2018, 224: 3~7. doi: 10.1016/j.elspec.2017.09.012
-
[9]
Zhang F, Song H, Song H, et al. J. Taiwan. Inst. Chem. E, 2016, 65: 558~564. doi: 10.1016/j.jtice.2016.05.048
-
[10]
Wei Q, Liu X, Zhou Y, et al. Catal. Today, 2020, 353: 39~46. doi: 10.1016/j.cattod.2019.10.032
-
[11]
Rodríguez-Aguado E, Infantes-Molina A, Ballesteros-Plata D, et al. Catal. Today, 2020, 349: 117~127. doi: 10.1016/j.cattod.2018.05.023
-
[12]
Li S, Zhang Q, Sun J, et al. Mater. Today Energy, 2020, 17: 100464. doi: 10.1016/j.mtener.2020.100464
-
[13]
Cheng L, Zhang D, Fan J, et al. Appl. Catal. A, 2020, 590: 117336. doi: 10.1016/j.apcata.2019.117336
-
[14]
Fan M, Chen Y, Xie Y, et al. Adv. Funct. Mater., 2016, 26(28): 5019~5027. doi: 10.1002/adfm.201601323
-
[15]
Durai L, Gopalakrishnan A, Badhulika S. J. Electroanal. Chem., 2020, 861: 113937. doi: 10.1016/j.jelechem.2020.113937
-
[16]
Xiao W, Zhang L, Bukhvalov D, et al. Nano Energy, 2020, 70: 104445. doi: 10.1016/j.nanoen.2020.104445
-
[17]
Oyama S T. J. Catal., 2003, 216(1-2): 343~352. doi: 10.1016/S0021-9517(02)00069-6
-
[18]
Oyama S T, Gott T, Zhao H, et al. Catal. Today, 2009, 143(1-2): 94~107. doi: 10.1016/j.cattod.2008.09.019
-
[19]
Gao L, Chen S, Zhang H, et al. Int. J. Hydrogen Energ., 2018, 43(30): 13904~13910. doi: 10.1016/j.ijhydene.2018.01.040
-
[20]
Gao Z, Gao Q, Liu Z, et al. RSC Adv., 2016, 6(115): 114430~114435. doi: 10.1039/C6RA24186G
-
[21]
Yang Y, Li J, Lv G, et al. J. Alloy Compd., 2018, 745: 467~476. doi: 10.1016/j.jallcom.2018.02.156
-
[22]
Zhou Z, Shi X, Yin J, et al. Chem. Phys. Lett., 2020, 749: 137403. doi: 10.1016/j.cplett.2020.137403
-
[23]
Song L, Zhang S. Powder Technol., 2011, 208(3): 713~716. doi: 10.1016/j.powtec.2011.01.014
-
[24]
Tang D, Li T, Li C M. Int. J. Hydrogen. Energ., 2019, 44(3): 1720~1726. doi: 10.1016/j.ijhydene.2018.11.157
-
[25]
Galindo-Ortega Y I, Infantes-Molina A, Huirache-Acuña R, et al. Fuel Proc. Technol., 2020, 208: 106507. doi: 10.1016/j.fuproc.2020.106507
-
[26]
Yang M, Zhu W, Zhao R, et al. J. Solid State Chem., 2020, 288: 121456. doi: 10.1016/j.jssc.2020.121456
-
[27]
Ranganatha S, Munichandraiah N. Mater. Chem. Phys., 2019, 224: 124~128. doi: 10.1016/j.matchemphys.2018.12.011
-
[28]
Song H, Gong J, Jiang N, et al. J. Fuel Chem. Technol., 2016, 44(5): 557~563. doi: 10.1016/S1872-5813(16)30025-1
-
[29]
Wang Z, Li L, Liu M, et al. J. Energy Chem., 2020, 48: 241~249. doi: 10.1016/j.jechem.2020.01.017
-
[30]
Jin C, Xu C, Chang W, et al. J. Alloy Compd., 2019, 803: 205~215. doi: 10.1016/j.jallcom.2019.06.252
-
[31]
黄晓昇, 赵吟霜, 吕功煊, 等. 分子催化, 2017, 31(3): 287~298. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGZS201903001.htm
-
[32]
Song L, Zhang S, Wei Q. Powder Technol., 2011, 212(2): 367~371. doi: 10.1016/j.powtec.2011.06.016
-
[33]
Dai X, Song H, Yan Z, et al. New J. Chem., 2018, 42: 19917~19923. doi: 10.1039/C8NJ04628J
-
[34]
Singh A, Chawla P, Jain S, et al. Physica E, 2017, 90: 175~182. doi: 10.1016/j.physe.2017.03.029
-
[35]
Ahluwalia D, Varshney A, Kumar S, et al. Inorg. Nano-Met. Chem., 2020, DOI: 10.1080/24701556.2020.1728551.
-
[36]
Jiang N, Jiang B, Wang J, et al. New. J. Chem., 2020, 44(20): 8379~8385. doi: 10.1039/D0NJ01106A
-
[37]
Usman M, Li D, Razzaq R, et al. J. Ind. Eng. Chem., 2015, 23: 21~26. doi: 10.1016/j.jiec.2014.08.033
-
[38]
Li G, Zhang X, Zhang H. Chem. Eng. J., 2020, 398: 125467. doi: 10.1016/j.cej.2020.125467
-
[39]
孟繁星, 遇治权, 景文文, 等. 高等学校化学学报, 2020, 41(4): 765~771. https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htm
-
[40]
Pan Z, Wang R, Li M, et al. J. Energy Chem., 2015, 24(1): 77~86. doi: 10.1016/S2095-4956(15)60287-X
-
[41]
Zhao S N, Song X Z, Song S Y, et al. Coord. Chem. Rev., 2017, 337: 80~96. doi: 10.1016/j.ccr.2017.02.010
-
[42]
Lee M H, Youn D H, Lee J S. Appl. Catal. A, 2020, 594: 117451. doi: 10.1016/j.apcata.2020.117451
-
[43]
Zhang J, Matsubara K, Yun G N, et al. Appl. Catal. A, 2017, 548: 39~46. doi: 10.1016/j.apcata.2017.06.009
-
[44]
Tong W, Xie Y, Luo H, et al. Chem. Eng. J., 2019, 378: 122187. doi: 10.1016/j.cej.2019.122187
-
[45]
Ma Z C, Chen Q D, Han B, et al. Langmuir, 2018, 34(20): 5712~5718. doi: 10.1021/acs.langmuir.7b04190
-
[46]
Zhang G, Xu Q, Liu Y, et al. Electrochim. Acta, 2020, 332: 135500. doi: 10.1016/j.electacta.2019.135500
-
[47]
Wang F, Qi X, Qin Z, et al. Int. J. Hydrogen Energ., 2020, 45(24): 13353~13364. doi: 10.1016/j.ijhydene.2020.03.064
-
[48]
Jing P, Wang Q, Wang B, et al. Ceram. Int., 2019, 45(1): 216~224. doi: 10.1016/j.ceramint.2018.09.154
-
[49]
Zhang X, Zhang L, Xu G, et al. J. Colloid Interf. Sci., 2020, 561: 23~31. doi: 10.1016/j.jcis.2019.11.112
-
[50]
d'Aquino A I, Danforth S J, Clinkingbead T R, et al. J. Catal., 2016, 335: 204~214. doi: 10.1016/j.jcat.2015.12.006
-
[51]
Song H, Yu Q, Chen Y, et al. Chin. J. Chem. Eng., 2018, 26(3): 540~544. doi: 10.1016/j.cjche.2017.09.001
-
[52]
Inocêncio C V M, de Souza P M, Rabelo-Neto R C, et al. Catal. Today, 2020. DOI: 10.1016/j.cattod.2020.07.077.
-
[53]
Ding X, Uddin W, Sheng H, et al. J. Alloy Compd., 2020, 814: 152332. doi: 10.1016/j.jallcom.2019.152332
-
[54]
Lin Y, Zhang M, Zhao L, et al. Appl. Surf. Sci., 2021, 536: 147952. doi: 10.1016/j.apsusc.2020.147952
-
[55]
Hu X, Yin Y, Liu W, et al. Chin. J. Catal., 2019, 40(7): 1085~1092. doi: 10.1016/S1872-2067(19)63299-7
-
[56]
Yang M, Jiang Y, Qu M, et al. Appl. Catal. B, 2020, 269: 118803. doi: 10.1016/j.apcatb.2020.118803
-
[57]
He L, Gong L, Gao M, et al. Electrochim. Acta., 2020, 337: 135799. doi: 10.1016/j.electacta.2020.135799
-
[58]
Xiao J, Zhang Z, Zhang Y, et al. Nano Energy, 2018, 51: 223~230. doi: 10.1016/j.nanoen.2018.06.040
-
[59]
Tang J Y, Yang D, Zhou W G, et al. J. Catal., 2019, 370: 79~87. doi: 10.1016/j.jcat.2018.12.009
-
[60]
Yuan Y J, Shen Z K, Song S, et al. ACS Catal., 2019, 9(9): 7801~7807. doi: 10.1021/acscatal.9b02274
-
[61]
Qin Z, Wang M, Li R, et al. Sci. China Mater., 2018, 61(6): 861~868. doi: 10.1007/s40843-017-9171-9
-
[62]
Man H, Tsang C, Li M M, et al. Appl. Catal. B, 2019, 242: 186~193. doi: 10.1016/j.apcatb.2018.09.103
-
[63]
Li N, Ding Y, Wu J, et al. ACS. Appl. Mater. Inter., 2019, 11(25): 22297~22306. doi: 10.1021/acsami.9b03965
-
[64]
Sun Z, Zhu M, Lv X, et al. Appl. Catal. B, 2019, 246: 330~336. doi: 10.1016/j.apcatb.2019.01.072
-
[65]
Hu B, Yuan J Y, Tian J Y, et al. J. Colloid Interf. Sci., 2018, 531: 148~159. doi: 10.1016/j.jcis.2018.07.037
-
[66]
Farahi E, Memarian N. Chem. Phys. Lett., 2019, 730: 478~484. doi: 10.1016/j.cplett.2019.06.041
-
[1]
-
-


 下载:
下载:







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 359
- 文章访问数: 12139
- HTML全文浏览量: 5378
