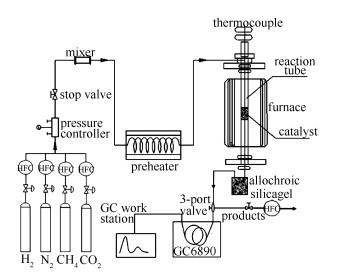
图 1.
催化剂评价装置示意图
Figure 1.
Schematic diagram of the catalyst evaluation device

CO2的大量排放已造成全球变暖,给人类乃至整个地球的生存带来严重的威胁和挑战。二十世纪初,Fischer和Tropsch发现了著名的CO2重整CH4反应 (CO2+CH4=2H2+2CO, CRM)。该反应不仅可获得重要的化工原料(合成气),还可同时利用CO2和CH4这两种典型的温室气体[1-3]。因此,CRM反应的研究与开发具有重要的工业和环境意义[4-6]。而且,CRM反应产物为H2/CO比接近于1的合成气,适宜于羰基合成和含氧有机化合物的合成[7]。所以,CO2-CH4重整反应是有效利用温室气体的一条可持续发展路线。
就CO2-CH4重整反应常用的Ni基催化剂而言,活性组分烧结和催化剂的表面积炭是阻碍该反应工业化应用的两大重要原因[8]。据报道,改变催化剂的制备条件、添加助剂、优化反应工艺条件均有助于改善活性组分的抗烧结能力,增强催化剂的抗积炭性能,从而提高反应的活性、选择性和稳定性[9-12]。
CO2-CH4催化重整体系中,CH4裂解(CH4=C+2H2)是主要的积炭反应,虽然CO2可发生消炭反应(CO2+C=2CO),但催化剂的表面积炭仍不可避免[13, 14]。Gadolalla等[15]以热力学分析为基础,研究了反应温度、反应压力和CO2/CH4物质的量比对积炭的影响,认为较高的反应温度和较低的压力以及较高的CO2/CH4物质的量比均可抑制积炭的发生。Wang等[16]研究了压力、进料比(CO2/CH4)与积炭产生的温度上限之间的关系。结果显示,进料比一定,催化剂表面产生积炭的温度上限与压力呈正相关。此外,反应压力不变,积炭的温度上限与进料比呈负相关。因此,可采用高温低压和增大CO2/CH4物质的量比的方法从热力学上抑制或防止积炭。
热力学计算结果显示[17],CO2和CH4按化学计量比1:1进料,反应温度高于等于645 ℃条件下,积炭的产生是不可避免的。若要抑制或避免积炭,有两条途径:一是提高反应温度,如当CO2/CH4进料比为1.2:1,温度高于等于1000 ℃,即能到达热力学的非积炭区;二是增大CO2与CH4的进料比,如当CO2与CH4物质的量比为3.7:1,温度高于等于800 ℃,即可避开Ni基催化剂上的积炭反应。然而,上述两种方法皆有不足之处。第一种方法所需温度高,能耗大,对装置材质要求苛刻;第二种方法增加CO2的进料比例,导致CO2利用率下降,后期的分离难度加大。从工业角度看,更期望在CO2与CH4物质的量比约为1:1的条件下进行反应。这就对催化剂本身和相应的工艺条件提出了更高的要求。
本研究采用水热沉积法,制备Ni质量分数约为8%的Ni-Al2O3催化剂。结合TPH表征,初步探讨操作条件,包括反应时间、反应温度、反应空速和CO2/CH4比例,对CO2-CH4重整反应积炭的影响规律。
采用水热沉积法,以Al2O3为载体制备催化剂(Al2O3制备方法详见文献[18])。以Ni(NO3)2·6H2O(A.R.,天津大茂化学试剂厂)为Ni源,NH2(CH2)2OH(A.R.,四川西陇化工有限公司)为沉淀剂。首先,称取4.32 g Ni(NO3)2·6H2O在室温下溶于装有5 mL蒸馏水的20 mL水热合成反应釜。然后,加入5 g载体Al2O3(40-60目),再以1 mL/min的速率滴加5 mL NH2(CH2)2OH,老化2 h。之后,在110 ℃下保持24 h,进行水热合成反应,反应结束将反应釜冷却至室温,用蒸馏水过滤洗涤釜内混合物至中性,除去残留沉淀剂,获得滤饼。最后,将滤饼于120 ℃下干燥3 h,800 ℃下焙烧3 h,即制得催化剂试样,记为Ni-Al2O3。
通过TPH测试催化剂表面的积炭量和积炭类型。在康塔公司Chem-BET pulsar TPR/TPD仪器上进行测试,催化剂用量50 mg,样品首先在He气氛中673 K下保持1 h进行前处理,然后切换为5%H2+95%Ar混合气,并以100 mL/min的气流进行TPH实验,以10 ℃/min的升温速率加热至1273 K。
热重主要用于测试催化剂表面的积炭量,采用美国TA公司的SDTQ600型仪器。条件:空气,流量100 mL/min;升温速率10 ℃/min,最高温度为1000 ℃。
采用日本Hitachi公司H-600型透射电子显微镜进行形貌观察,加速电压100 kV,粉末样品测试前先用无水乙醇超声分散,再滴到铜箔上,干燥后进行测试。
催化剂评价装置示意图见图 1,反应器为常压固定床不锈钢管。
反应管长400 mm,内径6 mm。催化剂粒径40-60目,装填量1.00 g。反应前,催化剂在800 ℃、氢气流量50 mL/min条件下还原3 h,而后切换至反应气体,实验条件为:CH4流量60 mL/min,CO2/CH4体积比为1,GHSV为14400 h-1。尾气由气相色谱仪(北京普瑞GC6890,ParapakQ柱,TCD检测器)在线检测。转化率和选择性按以下公式计算:
甲烷的转化率:
|
$ {x_{{\rm{C}}{{\rm{H}}_4}}} = \frac{{{n_{{\rm{C}}{{\rm{H}}_4}{\rm{, in }}}} - {n_{{\rm{C}}{{\rm{H}}_4}, {\rm{out}}}}}}{{{n_{{\rm{C}}{{\rm{H}}_4}, {\rm{out}}}}}} \times 100\% $ |
(1) |
CO2的转化率:
|
$ {x_{{\rm{C}}{{\rm{O}}_2}}} = \frac{{{n_{{\rm{C}}{{\rm{O}}_2}, {\rm{in}}}} - {n_{{\rm{C}}{{\rm{O}}_2}, {\rm{out}}}}}}{{{n_{{\rm{C}}{{\rm{O}}_2}, {\rm{in}}}}}} \times 100\% $ |
(2) |
H2的选择性:
|
$ {S_{{{\rm{H}}_2}}} = \frac{{{n_{{{\rm{H}}_2}, {\rm{ out }}}}}}{{{n_{{\rm{C}}{{\rm{H}}_4}, {\rm{ in }}}} - {n_{{\rm{C}}{{\rm{H}}_4}, {\rm{ out }}}}}} \times 100\% $ |
(3) |
设置反应时间分别为0.5、1、2、5和10 h,在800 ℃下考察了Ni-Al2O3催化剂上CO2-CH4重整反应的积炭情况。
为讨论CO2-CH4催化重整的反应性能,作为对比,在相同条件下,对固定床空反应管(未添加催化剂)的重整性能进行了考察,结果见图 2。
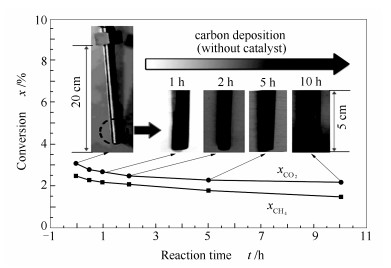
由图 2可知,在非催化条件下,CO2转化率不到3%,CH4转化率不到2%,即虽然实验条件下CO2-CH4重整反应依然可以进行,但转化率很低; 另一方面,Wang等[6]和Stubl等[14]等根据CRM反应标准自由能与温度的关系,计算出当温度高于等于640 ℃,即可发生CRM反应。而本实验采用的800 ℃已超过上述热力学反应温度,使反应在非催化条件下就可以发生。因此,可以认为实验所使用的不锈钢反应管对CRM反应几乎没有活性。
积炭方面,反应1 h后,热电偶管表面呈暗灰色,光滑度降低,未见明显积炭;反应5 h后,颜色转为黑褐色,可见些许积炭;反应10 h后,热电偶管表面可见少量黑色积炭。故而,在本实验条件下,热电偶管表面有积炭产生,但积炭量很小。
值得说明的是,催化反应条件下,热电偶管系插入催化剂床层中部(见图 1),可能会因反应的迅速、高效和长时间发生,而沉积更多的碳物种。由此,催化反应过程中,热电偶管或反应管表面积炭的绝大部分均来自于催化反应积炭。即积炭样品的回收和表征分析,仅针对反应后的催化剂开展相关测试。
图 3为CO2-CH4重整反应物转化率随时间的变化。由图 3可知,CO2和CH4的初始转化率均很高,说明本实验所制备的Ni-Al2O3催化剂的初始活性高。其中,CO2的初始转化率高达99.7%,这是因为CO2与重整反应生成的H2发生了RWGS反应(CO2+H2=CO+H2O),导致其转化率接近100%;CH4的初始转化率为94.9%,低于CO2转化率,这同样与逆水煤气反应的发生有关。另外,随着反应时间的延长,CO2与CH4转化率均呈下降趋势。前5 h下降趋势较为明显,5 h后下降趋势减缓;至反应10 h,CO2与CH4转化率分别降至95.5%和92.1%。
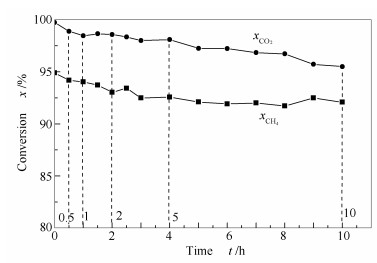
图 4为不同时间重整反应后试样的TPH谱图。由图 4可知,反应后的催化剂存在明显的表面氢化峰。据报道[18],200-350 ℃为第一类氢化峰,归属为无定型的α类型的碳物种,该碳物种是消炭反应的活性中间物(CO2+C=2CO),这也是涉碳反应所希望的积炭类型,此类型碳在高温下易转变成活性稍低的β类型的碳物种;350-500 ℃为第二类氢化峰,归属为β1类型的碳物种,未与载体形成较强的相互作用,其容易沉积在催化剂孔道,或者进入催化剂晶格形成碳纳米管或丝状炭,在较高温度下具有消炭反应活性,但高温下长时间聚集易转变成惰性的γ碳物种。500-700 ℃为第三类氢化峰,归属为γ类型的碳物种,如石墨炭,其活性低于α炭和β炭,是导致催化剂积炭不可逆失活的重要原因。文献还显示[19],在Ni-CaO-ZrO2催化剂表面发生CRM反应1 h后,程序升温加氢反应表征(TPH)发现,800 ℃左右出现的积炭氢化峰归属于β2类型的碳物种,即第四类氢化峰。因催化剂本身的金属-载体强相互作用和载体ZrO2的强氧化还原性能产生的电子效应,使该物种植入(encapsulate)载体内部,与载体形成强烈的相互作用,很难通过加氢反应而移除。
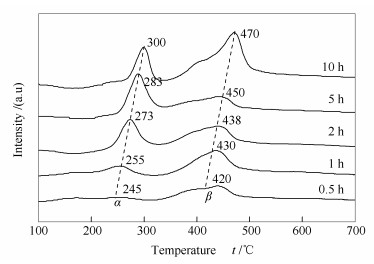
由图 4可知,在Ni-Al2O3催化剂上的CO2-CH4重整反应过程中,生成的碳物种表面氢化峰均在200-500 ℃,即主要以无定型炭Cα和丝状炭Cβ1类型存在[20, 21]。而在600和800 ℃左右未发现明显的氢化峰。故可认为,在10 h范围内,CO2-CH4重整反应几乎未形成Cγ和Cβ2类型的碳物种。
由图 4还可知,随重整反应时间的延长,Cα和Cβ两种积炭的氢化峰峰温均向高温方向移动,且峰面积明显增大。Cα的氢化峰温由245 ℃升高至300 ℃,Cβ的氢化峰温由420 ℃升高至470 ℃。说明随着反应时间的延长,积炭物种的类型可能会发生改变。可以推测,当反应时间继续延长,就有可能出现Cγ碳物种。另外,Cα和Cβ的氢化峰面积也呈增加趋势,表明反应时间越长,积炭量越大。但峰面积并未随反应时间的延长而成比例增大,说明积炭量亦未成比例增加。因此,催化剂表面很可能同时进行着积炭反应(CH4=C+2H2)和消炭反应(C+CO2=2CO)。
图 5为催化剂反应10 h后表面积炭实物图。由图 5可知,重整反应10 h后,热电偶管(插入催化剂床层部分)周围沉积了较厚的一层絮状炭(图 5(a)),催化剂表面由反应前的暗绿色变为反应后的纯黑色(图 5(b)),用普通白纸擦拭即可留下炭痕(图 5(c))。为进一步表征催化剂表面积炭量所占的比例和积炭类型,对反应后的催化剂进行了热重分析,见图 6。

(a): carbon deposited on the thermocouple tube;
(b): carbon on the Ni-Al2O3 catalyst;
(c): carbon on the wiping paper from thermocouple tube
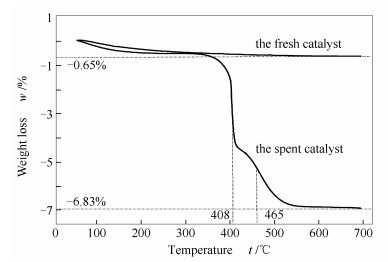
由图 6可知,新鲜催化剂失重率仅为0.65%,为本身所吸附的小分子气体脱附所致,这些小分子包括H2O、N2、O2等常见气体。因新鲜催化剂在保存和使用过程中,不可避免暴露于空气气氛,其较大的比表面积(200 m2/g左右,文献[18])使得对小分子的吸附量大于一般固体颗粒。
由图 6还可知,经重整反应积炭后的催化剂失重率为6.83%,扣除新鲜催化剂参比,为6.18%,其导致催化剂性能降低的原因有两个方面。一方面,积炭覆盖部分甚至全部活性中心,使重整反应无法正常进行,或者明显降低反应转化率,导致出口气体无法满足下游要求;另一方面,积炭堵塞催化剂孔道,导致反应气体无法进入催化剂的内表面(催化剂的比表面积以内表面为主),从而阻碍催化作用的发挥,影响正常生产。
另外,催化剂积炭的氧化脱除温度主要发生在408和465 ℃,说明积炭类型为无定型炭或丝状炭,两者的大量积累均会覆盖催化剂的活性中心或堵塞催化剂孔道,进而影响催化剂的性能。无定型炭或丝状炭可通过消炭反应(如:C+O2=CO2,俗称烧炭)得以有效去除。根据TG曲线,烧炭温度高于550 ℃即可达到有效除炭的目的,使得催化剂得到高效再生。另外,若进一步延长反应时间,上述两种积炭可能会转化为石墨炭,此类积炭很难通过消炭反应而去除,催化剂的再生极为困难。
图 7为新鲜催化剂和不同反应时间催化剂的TEM形貌结构照片。

(a): fresh; (b): 0.5 h; (c): 1 h; (d): 2 h; (e): 5 h; (f): 10 h
由图 7可知,新鲜催化剂表面未发现明显的积炭点(无定型炭)或碳纤维。随反应时间延长,催化剂表面开始出现纤维状积炭;反应2 h后,纤维状积炭开始累积增加,特别是反应5 h后,催化剂表面(图 7(e))可以观察到具有较多直径较大的纤维状或棒状形貌,这种形貌相互交织。随反应时间的进一步延长,纤维状积炭数量明显增多,而碳纤维的直径明显减小,这可能是在CO2-CH4重整反应中,消炭反应(C+CO2=2CO)的发生所致。另外,因催化剂在反应中承受了一定的压力,床层压降也随着积炭量的增加而增大,也可能引起纤维状积炭直径的减小。
在反应温度分别为650、700、750、800和850 ℃条件下,考察了Ni-Al2O3催化剂上CO2-CH4重整反应的积炭情况。
图 8为不同反应温度下试样的活性评价结果。由图 8可知,反应温度为650和700 ℃时,CH4和CO2转化率均较低,其中,700 ℃下两者转化率分别为60%和70%左右。反应温度为800和850 ℃时,CH4和CO2转化率均较高,其中,800 ℃两者转化率分别为85%和90%左右。显然,反应温度升高,催化剂活性得到明显提高,且750 ℃以后增加的趋势减缓。表明该反应是一个强吸热反应,这与重整反应的热力学本质一致(△H>0,吸热反应),即:
|
$ \begin{array}{*{20}{l}} {{\rm{C}}{{\rm{H}}_4}({\rm{g}}) + {\rm{C}}{{\rm{O}}_2}({\rm{g}}) = 2{\rm{CO}}({\rm{g}}) + 2{{\rm{H}}_2}({\rm{g}})}\\ {(\Delta H = 24{\rm{kJ}}/{\rm{mol}})} \end{array} $ |
(4) |
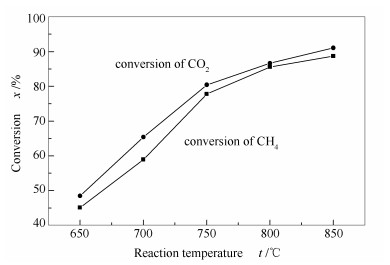
另外,当反应温度高于800 ℃,试样活性已达到一个较高值,如CH4和CO2的转化率在850 ℃反应温度下均高达90%以上。由不同反应温度下CH4的平衡转化率数据可知,反应温度升高,CH4转化率逐渐增大,并且在750 ℃以下这种升高幅度更大;而高于750 ℃,这种升高的趋势减缓,即反应转化率均接近于平衡转化率[22]。
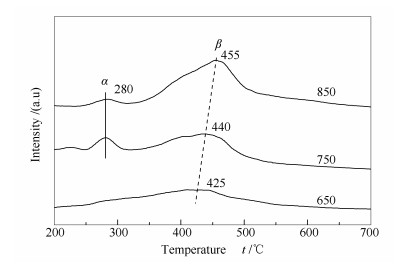
图 9为不同温度下重整反应后试样表面积炭的TPH表征结果。由图 9可知,各试样均存在低温和高温两个氢化峰,对应于两种类型的积炭物种。低温氢化峰在280 ℃左右,即无定型炭(α炭);高温氢化峰在450 ℃左右,即丝状炭(β炭)。上述两种类型的积炭物种均具有反应活性,可与CO2发生消炭反应。随着反应温度升高,积炭的高温氢化峰有向高温方向移动的趋势。反应温度从650 ℃升高到850 ℃,高温氢化峰的峰温从425 ℃升高到455 ℃,这是因为在高温条件下,丝状碳易向石墨炭转化,氢化反应需要在较高温度下方能进行[23]。
在空速分别为9600、12000、14400、16800和19200 h-1条件下,考察了Ni-Al2O3催化剂上CO2-CH4重整反应的积炭情况。
图 10为重整反应活性和选择性随空速的变化规律。
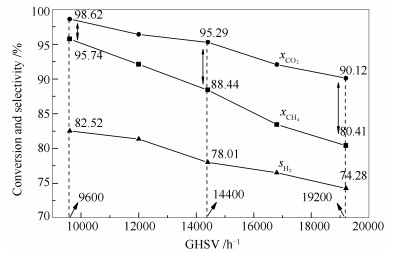
由图 10可知,随反应空速增加,CH4和CO2转化率出现明显的下降趋势。空速从9600 h-1增加到19200 h-1,CH4转化率从95.74%降至80.41%,降低了16.01%;CO2转化率从98.62%降至90.12%,降低了8.62%。因此,空速对催化剂活性影响较大。这可解释为,空速增加,反应气体在催化剂表面的停留时间缩短,部分反应物分子未及时扩散进入催化剂的孔道,导致单位时间内参与反应的分子数减少。
由图 10还可知,CH4转化率始终低于CO2转化率。如前所述,这是由于RWGS反应的存在引起。值得一提的是,随着反应空速增加,CH4转化率与CO2转化率的差距变大,从9600 h-1的2.88个百分点到14400 h-1的6.85个百分点,再到19200 h-1的9.71个百分点。这意味着CH4和CO2在催化反应体系中的扩散速率有差异。在低空速下,由于反应气体停留时间较长,CH4和CO2均有足够的时间扩散到催化剂的活性组分表面参与反应,两者转化率的差异仅由化学反应,如逆水煤气反应等因素造成;而在较高空速下,反应气体停留时间短,CH4和CO2没有足够的时间扩散到活性组分表面,这种情况下扩散速率较大的气体就更容易参与反应。从评价结果来看,高空速下CO2转化率与CH4转化率具有高达9.71个百分点的差异,这很可能是因为CO2在本体系条件下的扩散速率高于CH4所致。
图 11为不同空速下重整反应后试样的TPH谱图。
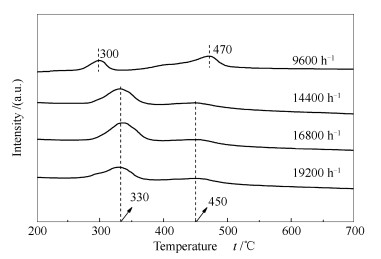
由图 11可知,反应后各催化剂表面均存在两种积炭类型,分别对应于330 ℃左右的α炭和450 ℃左右的β炭。在反应空速较低时,如本实验的9600 h-1,催化剂表面积炭的低温峰峰温较低,为300 ℃左右;高温峰峰温较高,在470 ℃左右。这可解释为,低空速下,CH4和CO2转化率较高,积炭量也较大,部分积炭已进入催化剂的微孔,氢化温度较高;部分积炭沉积在催化剂的外表面,氢化温度相对较低。而在反应空速较高时,反应气体快速通过催化剂孔道,并在孔道壁面反应产生积炭。这部分积炭未及时进入催化剂微孔,导致高温氢化峰的峰温和峰面积均相对较低;但所积之炭又沉积在催化剂的内孔道壁面,因而低温氢化峰相对较高[24, 25]。
在CO2/H4比分别为0.5:1、0.7:1、1:1、1.4:1和2:1条件下,考察了Ni-Al2O3催化剂上CO2-CH4重整反应的积炭情况。
图 12为不同CO2/CH4比例下试样重整反应的活性评价结果。由图 12可知,随着CO2/CH4比例的增加,CH4转化率升高,从0.5:1的60%左右升高到2:1条件下的98%以上;而CO2转化率降低,从0.5:1的98%以上下降到2:1条件下的70%左右。值得指出的是,在CO2/CH4比例为1时,CH4和CO2的转化率分别在85%和88%左右,仅相差3个百分点。当CO2/CH4比例低于1时,CH4过量,因而CO2转化率较高;当CO2/CH4比例大于1时,CO2过量,因而CH4转化率较高。
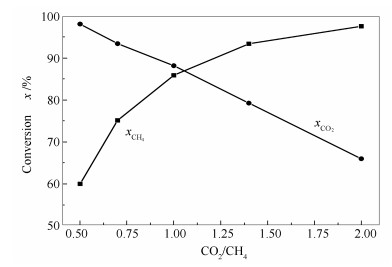
另外,按CO2-CH4重整反应方程式(CO2+CH4=2CO+2H2)计,CO2/CH4比例为0.5:1时,CH4的理论转化率为50%,而实验结果却在60%左右,这是因为反应温度为800 ℃,CH4会发生裂解反应(CH4=C+2H2)生成大量的H2,并伴有积炭的形成;另一方面,CO2/CH4比为2:1时,CO2的理论转化率为50%,而实验结果却在65%左右,这是因为副反应,即逆水煤气反应(CO2+H2=H2O+CO)的存在,导致CO2转化率高于理论值。
如前所述,联系CO2的消炭反应(CO2+C=2CO),可通过提高CO2的比例抑制积炭的形成,但CO2比例的提高又会促进副反应的发生。因此,关于CO2/CH4比例的确定需结合各方面的因素综合考虑。
图 13为不同CO2/CH4比例下重整反应后试样的TPH谱图。由图 13可知,除CO2/CH4为2:1条件下,试样表面积炭仅含有一个较小的氢化峰外,其他比例下试样表面均含有低温和高温两个氢化峰。随着CO2/CH4比例的增加,低温和高温氢化峰峰面积逐渐减小,说明催化剂表面积炭量逐渐降低;且高温氢化峰的减小程度更大,表明活性较低的β炭因CO2比例的提高显著减少。
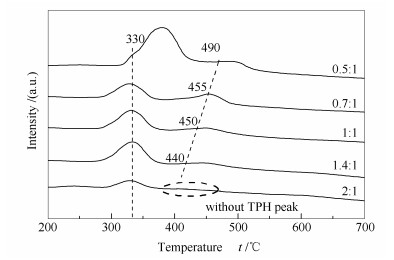
再者,高温氢化峰有向低温方向移动的趋势,从CO2/CH4为0.5:1的490 ℃降低到1.4:1的440 ℃,甚至到2:1时未发现高温氢化峰,说明CO2的大量加入抑制了非活性炭的形成。另外,在CO2/CH4为0.5:1条件下,催化剂表面积炭的低温氢化峰峰温高达380 ℃,明显高于其他条件下的峰温,意味着CO2比例较低时,CH4裂解产生的积炭易向非活性炭转变。上述结果说明,CO2的加入对抑制催化剂表面积炭和防止活性积炭向非活性积炭转变具有重要作用。因此,可通过提高原料气中CO2的比例,达到抑制积炭、提高催化剂稳定性的目的。
表面积炭是导致催化剂重整反应失活的重要原因。随反应时间延长,催化剂表面积炭量越多,但并未成比例增加。相对而言,反应温度和空速对催化剂表面积炭有一定影响,且空速的影响更大。另外,由于CO2消炭反应的存在,CO2/CH4比例对表面积炭的影响很大。结果显示,CO2/CH4比例太低,将无法达到明显抑制积炭的效果;CO2/CH4比例增加,积炭将得到有效抑制;而CO2/CH4比例太高,CO2在产物中的分离和回收再利用费用将明显增加。因此,CO2/CH4比例的确定需要结合多方面的因素综合分析。
MILICH L. The role of methane in global warming:Where might mitigation strategies be focused?[J]. Global Environ Change, 1999, 9(3): 179-201. doi: 10.1016/S0959-3780(98)00037-5
LAOSIRIPOJANA N, ASSABUMRUNGRAT S. Catalytic dry reforming of methane over high surface area ceria[J]. Appl Catal B:Environ, 2005, 60(1): 107-116.
THERDTHIANWONG S, THERDTHIANWON A, SIANGCHIN C, YONGPRAPAT S. Synthesis gas production from dry reforming of methane over Ni/Al2O3 stabilized by ZrO2[J]. Int J Hydrogen Energy, 2008, 33(3): 991-999. doi: 10.1016/j.ijhydene.2007.11.029
OYAMA S T, HACARLIOGLU P, GU Y F, LEE D. Dry reforming of methane has no future for hydrogen production:Comparison with steam reforming at high pressure in standard and membrane reactors[J]. Int J Hydrogen Energy, 2012, 37(13): 10444-10450. doi: 10.1016/j.ijhydene.2011.09.149
SUN H J, HUANG J, WANG H, ZHANG J G. CO2 reforming of CH4 over xerogel Ni-Ti and Ni-Ti-Al catalysts[J]. Ind Eng Chem Res, 2007, 46(13): 4444-4450. doi: 10.1021/ie070049e
WANG S B, LU G Q. Carbon dioxide reforming of methane to produce synthesis gas over metal-supported catalysts:State of the art[J]. Energy Fuels, 1996, 10: 896-904. doi: 10.1021/ef950227t
KOLESNICHENKO N V, GORYAINOVA T I, BIRYUKOVA E N, YASHINA O V, KHADZHIEV S N. Synthesis of lower olefins from dimethyl ether in the presence of zeolite catalysts modified with rhodium compounds[J]. Pet Chem, 2011, 51(1): 55-60.
GORYAINOVA T I, BIRYUKOVA E N, KOLESNICHENKO N V, KHADZHIEV S N. Study of magnesium-containing zeolite catalysts for the synthesis of lower olefins from dimethyl ether[J]. Pet Chem, 2011, 51(3): 169-173.
JIN L J, LI Y, LIN P, HU H Q. CO2 reforming of methane on Ni/γ-Al2O3 catalyst prepared by dielectric barrier discharge hydrogen plasma[J]. Int J Hydrogen Energy, 2014, 39(11): 5756-5763. doi: 10.1016/j.ijhydene.2014.01.171
JABBOUR K, EL HASSAN N, CASALE S, ESTEPHANE J, EL ZAKHEM H. Promotional effect of Ru on the activity and stability of Co/SBA-15 catalysts in dry reforming of methane[J]. Int J Hydrogen Energy, 2014, 39(15): 7780-7787. doi: 10.1016/j.ijhydene.2014.03.040
MOLINA R, PONCELET G. α-alumina-supported nickel catalysts prepared from nickel acetylacetonate:A TPR study[J]. J Catal, 1998, 173: 257-267. doi: 10.1006/jcat.1997.1931
YANG R C, LI X G, WU J S, ZHANG X, ZHANG Z H, CHENG Y F, GUO J T. Hydrotreating of crude 2-ethylhexanol over Ni/Al2O3 catalysts:Surface Ni species-catalytic activity correlation[J]. Appl Catal A:Gen, 2009, 368: 105-112. doi: 10.1016/j.apcata.2009.08.021
SONG K, LU M M, XU S P, CHEN C Q, ZH AN, Y Y, LI D L, AU C, JIANG L L, TOMISHIGE K. Effect of alloy composition on catalytic performance and coke-resistance property of Ni-Cu/Mg(Al)O catalysts for dry reforming of methane[J]. Appl Catal B:Environ, 2018, 239: 324-333. doi: 10.1016/j.apcatb.2018.08.023
STUBL D R, PROPHET H. JANAF Thermachemical Tables, NSRDS-NBS 37, Washington D.C, 1971.
GADDALLA A M, SOMMER M E. Carbon dioxide reforming of methane on nickel catalysts[J]. Chem Eng Sci, 1989, 44(12): 2825-2829. doi: 10.1016/0009-2509(89)85092-4
WANG H Y, RUCKENSTEIN E. Carbon dioxide reforming of methane to synthesis gas over supported rhodium catalysts:the effect of support[J]. Appl Catal A:Gen, 2000, 204(1): 143-152. doi: 10.1016/S0926-860X(00)00547-0
CHEN Q J, ZHANG J, PAN B R, KONG W B, CHEN Y Y, ZHANG W L, SUN Y H. Temperature-dependent anti-coking behaviors of highly stable Ni-CaO-ZrO2 nanocomposite catalysts for CO2 reforming of methane[J]. Chem Eng J, 2017, 320: 63-73. doi: 10.1016/j.cej.2017.03.029
MO, MA, LIU, LIU, AISHA·NULAHONG. Preparation of porous Al2O3 by template method and its application in Ni-based catalyst for CH4/CO2 reforming to produce syngas[J]. Int J Hydrogen Energy, 2015, 40(46): 16147-16158. doi: 10.1016/j.ijhydene.2015.09.149
WANG C Z, SUN N N, WEI W, ZHANG Y X. Carbon intermediates during CO2 reforming of methane over Ni-CaO-ZrO2 catalysts:A temperature-programmed surface reaction study[J]. Int J Hydrogen Energy, 2016, 41(42): 19014-19024. doi: 10.1016/j.ijhydene.2016.08.128
BODROV I M, APELBAUM L O. Reaction kinetics of methane and carbon dioxide on a nickel surface[J]. Kinet Catal, 1967, 8(2): 379.
LI D L, XU S P, SONG K, CHEN C Q, ZHAN Y Y, JIANG L L. Hydrotalcite-derived Co/Mg(Al)O as a stable and coke-resistant catalyst for low-temperature carbon dioxide reforming of methane[J]. Appl Catal A:Gen, 2018, 552: 21-29. doi: 10.1016/j.apcata.2017.12.022
DAI C Y, ZHANG S H, ZHANG A F, SONG C S, SHI C, GUO X W. Hollow zeolite encapsulated Ni-Pt bimetals for sintering and coking resistant dry reforming of methane[J]. J Mater Chem A, 2015, 3(32): 16461-16468. doi: 10.1039/C5TA03565A
WANG R, XU H Y, LIU X B, GE Q J, LI W Z. Role of redox couples of Rh0/Rhδ+ and Ce4+/Ce3+ in CH4/CO2 reforming over Rh-CeO2/Al2O3 catalyst[J]. Appl Catal A:Gen, 2006, 305(2): 204-210. doi: 10.1016/j.apcata.2006.03.021
KIM J H, SUH D J, PARK T J, KIM K L. Effect of metal particle size on coking during CO2 reforming of CH4 over Ni-alumina aerogel catalysts[J]. Appl Catal A:Gen, 2000, 197(2): 191-200. doi: 10.1016/S0926-860X(99)00487-1
SOUZA M M V M, ARANDA D A G, SCHMAL M. Coke formation on Pt/ZrO2/Al2O3 catalysts during CH4 reforming with CO2[J]. Ind Eng Chem Res, 2002, 41(18): 4681-4685. doi: 10.1021/ie010970a

图 2 固定床空反应管CO2-CH4重整性能
Figure 2 Conversions of CO2 and CH4 for the CO2-CH4 reforming in the empty fixed bed reactor without loading any catalyst at 800 ℃, 0.1 MPa, GHSV = 14400 h-1, and CO2/CH4 =1

图 3 重整反应CO2和CH4转化率随时间的变化
Figure 3 Conversions of CO2 and CH4 for the CO2-CH4 reforming over the Ni-Al2O3 catalyst for different reaction times at 800 ℃, 0.1 MPa, GHSV = 14400 h-1, and CO2/CH4 =1

图 4 不同时间重整反应试样的TPH谱图
Figure 4 TPH profiles of various spent Ni-Al2O3 catalysts after carrying out the CO2-CH4 reforming reaction for different times

图 5 催化剂反应10 h后表面积炭实物示意图
Figure 5 Photos of the carbon deposited after carrying out the CO2-CH4 reforming reaction for 10 h
(a): carbon deposited on the thermocouple tube;
(b): carbon on the Ni-Al2O3 catalyst;
(c): carbon on the wiping paper from thermocouple tube

图 6 反应10 h后催化剂的热重分析曲线
Figure 6 TG curves of the fresh Ni-Al2O3 catalyst and the spent one after carrying out the CO2-CH4 reforming for 10 h

图 7 不同反应时间下催化剂表面积炭的TEM照片
Figure 7 TEM images of various spent Ni-Al2O3 catalysts after carrying out the CO2-CH4 reforming reaction for different times
(a): fresh; (b): 0.5 h; (c): 1 h; (d): 2 h; (e): 5 h; (f): 10 h

图 8 不同温度下重整反应的转化率随温度的变化
Figure 8 Conversions of CO2 and CH4 for the CO2-CH4 reforming over the Ni-Al2O3 catalyst under different temperatures after reaction for 10 h at 0.1 MPa, GHSV = 14400 h-1, and CO2/CH4 =1

图 9 不同温度下重整反应后试样的TPH谱图
Figure 9 TPH profiles of the spent Ni-Al2O3 catalysts after carrying out the CO2-CH4 reforming reaction under different temperatures

图 10 不同空速下重整反应转化率和选择性随空速的变化
Figure 10 Conversions of CO2 and CH4 for the CO2-CH4 reforming over the Ni-Al2O3 catalyst under different GHSVs after reaction for 10 h at 800 ℃, 0.1 MPa, and CO2/CH4 =1

图 11 不同空速下重整反应后试样的TPH谱图
Figure 11 TPH profiles of the spent Ni-Al2O3 catalysts after carrying out the CO2-CH4 reforming reaction under different GHSVs

图 12 不同CO2/CH4比例下试样重整反应的活性评价
Figure 12 Conversions of CO2 and CH4 for the CO2-CH4 reforming over the Ni-Al2O3 catalyst under different CO2/CH4 ratios after reaction for 10 h at 800 ℃, 0.1 MPa, and GHSV=14400 h-1

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
