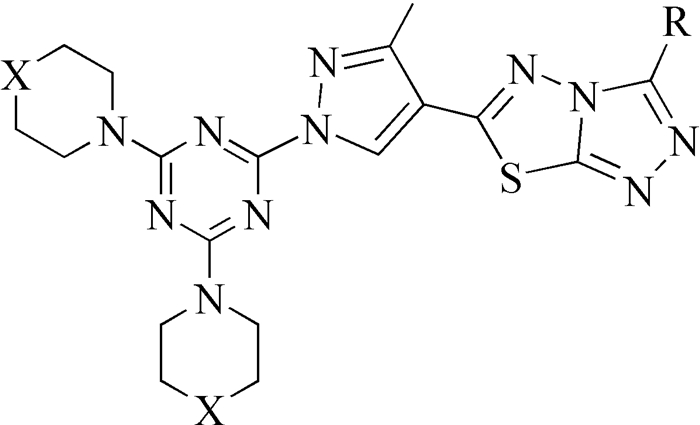
图式 1.
三唑并噻二唑衍生物的母体结构
Scheme 1.
Basic structure of novel triazol-ethiadiazole derivatives

蛋白酪氨酸磷酸酯酶1B(Protein tyrosine phosphatase 1B,PTP1B)属于蛋白酪氨酸磷酸酯酶(Protein tyrosine phosphatases,PTPs),具有促进细胞有丝分裂作用,或在胰岛素信号转导通路中起着重要的负调控作用。PTP1B还与肿瘤、自身免疫和心血管等许多疾病密切相关。因此探究类药性好和活性优良的PTP1B抑制剂,无疑对筛选出抗肿瘤和抗糖尿病先导药物具有重要意义。张成路等[1]根据亚结构拼接和活性叠加原理,以具有优异药理活性的吡唑杂环为核心,连接1, 3, 5-三嗪杂环,并引入三唑并噻二唑稠环,设计合成了21个新型1, 3, 5-三嗪-1H-吡唑-三唑并噻二唑衍生物,简称“三唑并噻二唑衍生物”。他们以齐墩果酸为阳性对照物,测试上述化合物对PTP1B的抑制活性,以“IP”表示,单位为μg·cm-3。
物质定量构效关系(QSAR)[2~6]是描述分子结构和分子某种生物活性之间的数学模型。其基本假设是化合物的分子结构决定其生物活性,而生物活性只是其结构的反映。因此,反映化合物分子结构的数据与其生物活性必然存在某种程度上的相关。QSAR一般分为二维和三维构效关系,即2D/3D-QSAR。其中2D-QSAR已被许多学者用来研究物质的各种性能[2~10]。3D-QSAR是引入了分子三维结构信息进行定量构效关系的研究方法,比2D-QSAR有更加明确的物理意义和更丰富的信息量。应用最广泛的3D-QSAR是CoMFA[11~17],即比较分子场方法。CoMFA主要是应用探针原子进行立体场和静电场的场分析,得出三维等值线图,从而得到3D-QSAR模型。此方法主要用于药物的构效关系和分子设计研究。本文基于CoMFA方法,建立21个三唑并噻二唑衍生物对PTP1B抑制活性的3D-QSAR模型,以探讨影响抑制活性的微观结构因素,揭示抑制作用的分子机理。
张成路等[1]以具有优异药理活性的吡唑杂环为核心,设计合成21个新型三唑并噻二唑衍生物,其母体结构见图式 1。
其中,取代基X、R见表 1。考虑这些化合物对PTP1B的抑制作用,显然是以分子整体发挥作用,因此,令:
 下载:
导出CSV
下载:
导出CSV
| Compd. | R | X | IP.exp[1] | pMP.exp. | pMP.cal. |
| 1 | Ph | O | 6.12 | 4.938 | 5.002 |
| 2* | 4-MePh | O | 3.64 | 5.175 | 5.339 |
| 3 | 4-OHPh | O | 0.90 | 5.784 | 5.686 |
| 4 | 4-ClPh | O | 1.22 | 5.666 | 5.699 |
| 5 | 4-(1-pyrrolidine formyl) Ph | O | 6.98 | 4.954 | 4.992 |
| 6 | n-C5H11 | O | 1.19 | 5.645 | 5.633 |
| 7* | n-C11H23 | O | 4.01 | 5.182 | 5.585 |
| 8 | Ph | CH2 | 3.72 | 5.151 | 5.080 |
| 9 | 4-MePh | CH2 | 2.96 | 5.262 | 5.254 |
| 10 | 4-OHPh | CH2 | 0.67 | 5.909 | 5.839 |
| 11 | 4-ClPh | CH2 | 1.10 | 5.708 | 5.782 |
| 12 | 4-(1-pyrrolidine formyl) Ph | CH2 | 4.77 | 5.117 | 5.099 |
| 13* | n-C5H11 | CH2 | 1.02 | 5.708 | 5.604 |
| 14 | n-C11H23 | CH2 | 3.66 | 5.218 | 5.273 |
| 15 | Ph | 0 | 6.01 | 4.919 | 4.968 |
| 16 | 4-MePh | 0 | 3.42 | 5.176 | 5.145 |
| 17 | 4-OHPh | 0 | 0.87 | 5.772 | 5.844 |
| 18* | 4-ClPh | 0 | 1.16 | 5.663 | 5.549 |
| 19 | 4-(1-pyrrolidine formyl) Ph | 0 | 5.51 | 5.034 | 5.014 |
| 20 | n-C5H11 | 0 | 1.11 | 5.648 | 5.647 |
| 21 | n-C11H23 | 0 | 3.78 | 5.184 | 5.127 |
| 22* | 4-FPh | CH2 | 5.928 | ||
| 23* | 4-CNPh | CH2 | 6.292 | ||
| 24* | 4-CF3Ph | CH2 | 6.033 |
|
$ M_{\mathrm{p}}=I_{\mathrm{P}} / M_{\mathrm{r}} $ |
(1) |
式中,Mr为化合物的摩尔质量,MP的单位为μmol·dm-3。根据物理化学原理,化学平衡常数与组分的平衡浓度之间为对数关系。因此,取MP的负对数,即pMP用于建模。pMP的具体数值见表 1。
本文使用Tripos公司最新Sybyl-x2.1.1分子模拟软件建立CoMFA模型及3D-QSAR分析,包括SYBYL、Sketch、Minimize、Database-alignment以及CoMFA的QSAR方法等,各项参数除特别指明外均采用缺省值。
分子活性构象的确定是建立有效3D-QSAR模型的首要前提之一。一般采用分子的最低能量构象代替分子的药效构象,即活性构象。具体步骤:首先利用Sybyl-x2.1.1软件中的Sketch molecule模块构建24个三唑并噻二唑衍生物分子(含设计的3个分子)的初始三维结构,然后通过Minimize模块,选取Tripos力场,加Gasteiger-Huckel电荷,将最大迭代次数(Max. Iterations)定为1000,将Gradient降低到0.005;其余采用缺省值。以此对上述分子进行分子力学优化,并以获得的最低能量构象作为分子叠合的最终构象。
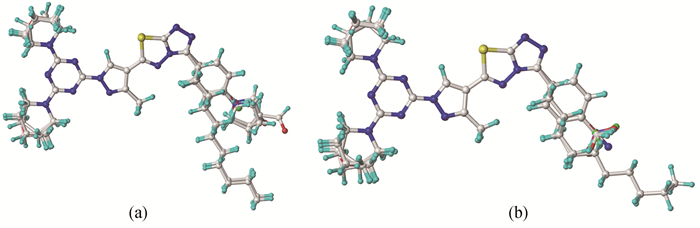
随机选取分子2、7、10、13、18及设计的3个分子为测试集(含模板分子10及表 1中带“*”分子,共8个分子),余下17个分子作为训练集(含模板分子10)。据CoMFA的研究方法,选用对PTP1B的抑制作用最大的化合物10为模板分子。基于公共骨架的叠合方法,即以图 1式中除去“R、X”以外的所有非氢原子为叠合的公共骨架进行叠合。分别运用Align database模块对上述训练集、测试集进行叠合,为使分子间相互重叠时的均方根偏差最小,尽量保证所有分子取向的一致性。训练集、测试集的叠合图见图 1。
用已叠合好的17个训练集分子建立CoMFA模型。采用Tripos标准力场,对叠合分子周围每个网格点上的立体场(Steric,S)及静电场(Electrostatic,E)予以计算,场能阈值(Cut off值)设为125.5 kJ·mol-1,其余各项参数均为系统默认值。在进行偏最小二乘法(Partial least squares,PLS)进行分析时,首先采用逐一剔除法(Leave-one-out,LOO)予以交叉验证,以获交叉验证系数Rcv2和最佳主成分数N。在CoMFA模型的PLS分析中,以统计指标为Rcv2衡量模型的预测能力。当Rcv2>0.3时,所建立模型在5%显著水平上有统计意义,其机会相关的可能性小于5%(该模型可信度为95%)[18]。然后通过非交叉验证进行回归分析,得到非交叉验证判定系数R2、标准偏差SD、统计方差比F,以及3D-QSAR模型。最后采用View CoMFA模块,以三维等势图直观反映化合物分子周边的立体场和静电场对PTP1B抑制作用的贡献。
训练集所建CoMFA的3D-QSAR模型:最佳主成分数N=7,交叉验证系数Rcv2=0.432>0.3,说明模型具有较好的预测能力。此外其传统判定系数R2为0.975,标准偏差SD为0.073、统计方差比F为49.403,这都证明模型具有良好的相关性、统计学稳定性。利用该模型对训练集的估算值及对测试集的预测值均见表 1的pMP.cal.,与相应实验值基本吻合,表明模型具有良好的预测能力。
图 2给出了以训练集中pMP最高的分子10为模板的CoMFA模型的三维等势图,分子周围不同颜色的块状图表示分子10周边的立体场和静电场对pMP的影响。图 2(a)为立体作用分布图,其中绿色表示在该区域增大取代基体积有利于提高化合物的抑制活性,黄色区域表示减小立体位阻对抑制活性有利。由立体场的空间分布可知,在4-位引入一个体积较大基团,在3-位引入小体积基团,有助于提高三唑并噻二唑衍生物对PTP1B抑制作用。对于分子1与2、8与9、15与16的3-位上均为H,而4-位均是前为H、后为CH3,所以,pMP顺序为1>2、8>9、15>16。
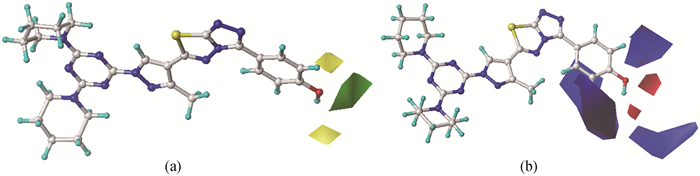
图 2(b)为化合物周围的静电场分布,蓝色表示在该区域增大基团正电性有利于化合物活性的提高,红色区域则表示引入带负电荷的基团对活性有利。由静电场的空间分布可知,在苯环3-位、5-位上引入正电性基团,在4-位引入一个负电性基团,有助于提高三唑并噻二唑衍生物对PTP1B抑制作用。显然,OH的负电性强于Cl,可与PTP1B形成氢键,增加与受体的亲和力和选择性,从而使化合物发挥优异的药理活性作用。对于分子3、10、17的4-位均是OH,而4、11、18的4-位均是Cl。因此,pMP具有如下顺序:3>4、10>11、17>18。
训练集的3D-QSAR模型给出模板分子10的周围立体场和静电场对pMP的贡献依次为59.2%和40.8%,显示立体作用大于静电作用。立体场一般对应于位阻及疏水作用,静电场通常包含氢键及配位作用。据此推测三唑并噻二唑衍生物分子与PTP1B之间主要是位阻、疏水、氢键及配位作用。如化合物6、13、20的pMP大于7、14、21,是因为后者R为C11H23,脂肪碳链过长,在与PTP1B受体相作用时,存在空间位阻或分子缠绕,阻碍了分子中活性位点与受体靶点的相互作用,使其抑制活性受到较大影响。由此可见,本文研究结果与文献[1]结论基本一致。
QSAR研究目的之一是依据所建模型中隐含的结构信息设计分子,并运用该模型去预测所设计分子的生物活性,为新型的、高活性分子的合成、生物学实验提供理论依据。根据3.2节的分析可知,在苯环4-位上引入体积较大的负电性基团,有利于增强三唑并噻二唑衍生物的抗肿瘤活性。据此设计3个化合物(见表 1中化合物22~24),3D-QSAR模型给出较大的pMP预测值,均优于文献中最佳的抑制活性。这些理论上优良的PTP1B抑制剂,尚需生物医学实验确认。
本文采用CoMFA方法研究三唑并噻二唑衍生物的三维定量构效关系,获得合理、可信、有良好预测性的3D-QSAR模型。所建CoMFA模型较好地解释了训练集分子抗肿瘤活性的差异,是因取代基团在整个分子上的立体场和静电场双重分布作用的不同。三唑并噻二唑衍生物分子与PTP1B之间主要是位阻、疏水、氢键及配位作用。据三唑并噻二唑衍生物分子周边立体场和静电场分布特征,设计了3个具有优于文献[1]中最佳抑制活性的分子,有待生物医学实验验证。
张成路, 李传银, 顾耀东等.有机化学, 2018, 38(5):1223~1232. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=yjhx201805023
张玲, 刘鹰翔, 赵钟祥等.化学通报, 2018, 81(2):148~157. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20170929002&flag=1
冯长君.徐州工程学院学报(自然科学报), 2018, 33(4):38~44. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=xzgcxyxb-zk201804007
M F Hassan, S G Adel, D T Anand et al. Eur. J. Med. Chem., 2018, 143(1):1524~1534.
C Wang, C J Feng. Chin. J. Struct. Chem., 2017, 36(10):1720~1728.
C Wang, C J Feng. Chin. J. Struct. Chem., 2018, 37(11):1679~1688.
冯长君.徐州工程学院学报(自然科学报), 2017, 32(3):22~27. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=xzgcxyxb-zk201703004
R J Qu, H X Liu, M B Feng et al. J. Chem. Engin. Data, 2012, 57(9):2442~2455. doi: 10.1021/je300407g
X L Zeng, R J Qu, M B Feng et al. Envir. Sci. Tech., 2016, 50(15):8128~8134. doi: 10.1021/acs.est.6b02682
H Liu, P Sun, H X Liu et al. Chemosphere, 2015, 135:182~188. doi: 10.1016/j.chemosphere.2015.04.036
冯长君.徐州工程学院学报(自然科学报), 2019, 34(2):20~25.
S Ahamad, A Islam, F Ahmad et al. Comput. Biol. Chem., 2019, 78:398~413. doi: 10.1016/j.compbiolchem.2018.12.017
李鸣建, 冯惠, 冯长君等.化学通报, 2015, 78(2):153~157. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20140611002&flag=1
W J Zhang, Z Wei, C Y Lin et al. J. Mol. Struct., 2019, 1186:11~22. doi: 10.1016/j.molstruc.2019.02.107
曹洪玉, 吴艳华, 任聪等.化学通报, 2018, 81(6):548~554. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20180116002&flag=1
H X Liu, J Q Shi, H Liu et al. Atmos. Environ., 2013, 77:840~845. doi: 10.1016/j.atmosenv.2013.05.068
R D Cramer, D E Patterson, J D Bunce. J. Am. Chem. Soc., 1988, 110(18):5959~5967. doi: 10.1021/ja00226a005
胡松青, 米思奇, 贾晓林等.高等学校化学学报, 2011, 32(10):2402~2409. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=gdxxhxxb201110032

图式 1 三唑并噻二唑衍生物的母体结构
Scheme 1 Basic structure of novel triazol-ethiadiazole derivatives

图 1 训练集(a)与测试集(b)的分子叠合图
Figure 1 The plots of all aligned compounds in the training(a) and test set(b)

图 2 CoMFA模型的立体场(a)与静电场(b)等势图
Figure 2 CoMFA contour maps (a) steric field; (b) electrostatic field
表 1 三唑并噻二唑衍生物的分子结构与抑制活性(pMP)
Table 1. The inhibitory activities (pMP) and molecular structures of novel triazol-ethiadiazole derivatives
| Compd. | R | X | IP.exp[1] | pMP.exp. | pMP.cal. |
| 1 | Ph | O | 6.12 | 4.938 | 5.002 |
| 2* | 4-MePh | O | 3.64 | 5.175 | 5.339 |
| 3 | 4-OHPh | O | 0.90 | 5.784 | 5.686 |
| 4 | 4-ClPh | O | 1.22 | 5.666 | 5.699 |
| 5 | 4-(1-pyrrolidine formyl) Ph | O | 6.98 | 4.954 | 4.992 |
| 6 | n-C5H11 | O | 1.19 | 5.645 | 5.633 |
| 7* | n-C11H23 | O | 4.01 | 5.182 | 5.585 |
| 8 | Ph | CH2 | 3.72 | 5.151 | 5.080 |
| 9 | 4-MePh | CH2 | 2.96 | 5.262 | 5.254 |
| 10 | 4-OHPh | CH2 | 0.67 | 5.909 | 5.839 |
| 11 | 4-ClPh | CH2 | 1.10 | 5.708 | 5.782 |
| 12 | 4-(1-pyrrolidine formyl) Ph | CH2 | 4.77 | 5.117 | 5.099 |
| 13* | n-C5H11 | CH2 | 1.02 | 5.708 | 5.604 |
| 14 | n-C11H23 | CH2 | 3.66 | 5.218 | 5.273 |
| 15 | Ph | 0 | 6.01 | 4.919 | 4.968 |
| 16 | 4-MePh | 0 | 3.42 | 5.176 | 5.145 |
| 17 | 4-OHPh | 0 | 0.87 | 5.772 | 5.844 |
| 18* | 4-ClPh | 0 | 1.16 | 5.663 | 5.549 |
| 19 | 4-(1-pyrrolidine formyl) Ph | 0 | 5.51 | 5.034 | 5.014 |
| 20 | n-C5H11 | 0 | 1.11 | 5.648 | 5.647 |
| 21 | n-C11H23 | 0 | 3.78 | 5.184 | 5.127 |
| 22* | 4-FPh | CH2 | 5.928 | ||
| 23* | 4-CNPh | CH2 | 6.292 | ||
| 24* | 4-CF3Ph | CH2 | 6.033 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们