 图 1
GPR40介导的信号通
Figure 1.
GPR40-mediated signaling pathways
图 1
GPR40介导的信号通
Figure 1.
GPR40-mediated signaling pathways
引用本文:
李鹤, 龙亚秋. 小分子GPR40激动剂作为抗Ⅱ型糖尿病候选药物的研究进展[J]. 有机化学,
2015, 36(4): 736-743.
doi:
10.6023/cjoc201511011

Citation: Li He, Long Yaqiu. Advances in Small-Molecule GPR40 Agonists for Treatment of Type 2 Diabetes Mellitus[J]. Chinese Journal of Organic Chemistry, 2015, 36(4): 736-743. doi: 10.6023/cjoc201511011
Citation: Li He, Long Yaqiu. Advances in Small-Molecule GPR40 Agonists for Treatment of Type 2 Diabetes Mellitus[J]. Chinese Journal of Organic Chemistry, 2015, 36(4): 736-743. doi: 10.6023/cjoc201511011
小分子GPR40激动剂作为抗Ⅱ型糖尿病候选药物的研究进展
摘要:
目前治疗Ⅱ型糖尿病的主要手段依然是口服或注射降糖药物以达到控制血糖的目的.虽然已有针对不同靶点开发的多种抗Ⅱ型糖尿病药物上市, 但是鉴于糖尿病治疗药物长期服用的高安全性要求以及庞大的患病人群, 研发新型安全有效的抗糖尿病药物仍然是当今药物化学研究的热点.GPR40是G-蛋白偶联受体家族中的一员, 激动后可以诱导葡萄糖依赖的胰岛素分泌.因为仅在血糖浓度过高时, GPR40激动剂才能促进胰岛素分泌, 所以针对该靶点开发降糖药物将极大地降低目前抗糖尿病药物低血糖副作用的风险.因此, GPR40已成为抗Ⅱ型糖尿病药物研发的前沿和热门靶点, 本文将围绕小分子GPR40激动剂的药效团模型, 即必需的苯丙酸核心骨架及其疏水末端和连接链的结构改造, 对近年来各大制药公司及研究机构报道的小分子GPR40激动剂的研发进展进行总结评述.
-
关键词:
- Ⅱ型糖尿病
- / GPR40
- / 激动剂
- / 葡萄糖依赖的胰岛素分泌
- / 苯丙酸
English
Advances in Small-Molecule GPR40 Agonists for Treatment of Type 2 Diabetes Mellitus
Abstract:
Currently, the majority of the chemotherapy for type 2 diabetes mellitus (T2DM) functions through glycemic control by administration of oral or injectable hypoglycemic drugs. Though a range of anti-diabetic drugs with different modes of action have been launched, there still remains a significant need for development of effective and highly safe anti-diabetic agents for the increasing diabetic population. GPR40 belongs to the GPCR family and the activation of GPR40 can amplify glucose-stimulated insulin secretion (GSIS). Due to the advantage of minimizing the hypoglycemia risk, GPR40 has drawn more and more attention and emerged as a promising new target for T2DM treatment. Therefore, the recent progress on the structural optimization and further development of small molecule GPR40 agonists as novel treatment for T2DM is reviewed, focused on those under clinical trials or at preclinical stage. A variety of small molecule GPR40 agonists were summarized from the literature and patents based on the pharmacophore model, including the critical phenylpropanoic acid core, the hydrophobic terminus and the linker. The structural features and advantage of different sources of GPR40 agonists were analyzed and highlighted.
-
Ⅱ型糖尿病是一种严重危害人类健康的代谢性疾病.据统计, 目前全球已有超过3亿的二型糖尿病患者[1].其主要特点表现为由于胰岛素抵抗导致的高血糖.血糖浓度过高将带来一系列并发症的风险, 如心血管系统疾病、糖尿病肾病、糖尿病足、失明等.目前治疗Ⅱ型糖尿病的主要手段就是通过口服或注射降糖药物控制患者的血糖浓度.由于Ⅱ型糖尿病患者需要长期服药控制血糖, 所以这对抗Ⅱ型糖尿病药物的安全性提出了更高的要求.虽然目前已批准上市了多种抗Ⅱ型糖尿病药物, 但是多数药物在降血糖的同时会伴有导致低血糖的副作用[2].此外, 一些药物还会引起胃肠道不适、水肿、骨折风险增高等副作用.因此, 开发安全性高且口服有效的新型抗Ⅱ型糖尿病药物一直是药物化学家努力的方向.
近几年来, 随着胰高血糖素样肽-1 (GLP-1)类似物和二肽基肽酶4 (DPP4)抑制剂的成功上市, 抗Ⅱ型糖尿病药物的开发主要集中在能够诱导葡萄糖依赖的胰岛素分泌的靶点上, 即药物仅在血糖浓度高时, 才会促进胰岛素分泌.这样就大大降低了产生低血糖的风险, 为开发更安全的抗Ⅱ型糖尿病药物提供了理论基础. GPR40便是具备了这样优秀特点的一个新靶点, 因此世界多家制药公司和研究机构对其予以了大量投入, 希望以其为靶点开发出更安全的新型抗Ⅱ型糖尿病药物.
GPR40, 又称为FFA1受体, 是GPCR家族中的一员, 主要分布于胰岛β细胞[3]和肠内分泌细胞I、K和L细胞[4].近期还有研究显示, GPR40在脑内也有分布, 其作用可能与疼痛调节有关[5].
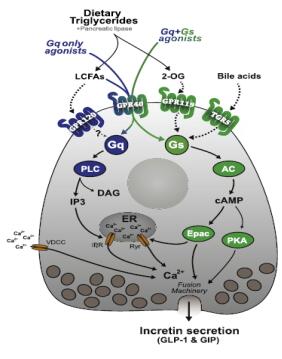
GPR40的内源性激动剂为中、长链脂肪酸, 如癸酸、软质酸、油酸、二十二碳六烯酸等[3].急性的脂肪酸激动GPR40可引发葡萄糖依赖的胰岛素分泌, 但是长期的高脂肪酸会导致胰岛β细胞损伤.内源性激动剂与GPR40结合后, 能够通过Gq/IP3通路, 导致钙流增加, 促进胰岛素分泌.最近的研究显示, 一些合成的GPR40激动剂如Amgen公司研发的AM 1638和AM 5262不仅能够激活Gq/IP3通路, 还能够激活Gs/cAMP通路, 从而导致GLP-1和GIP的释放(图 1)[6].
图 1
GPR40介导的信号通
Figure 1.
GPR40-mediated signaling pathways
目前报道的GPR40激动剂多为模块化分子, 其药效团模型大致可归纳为苯丙酸/丙酸的生物电子等排体、连接链和芳香片段三部分(图 2).其中羧基的β位常引入一些体积较小的取代基, 以减少β氧化, 提高化合物的代谢稳定性.所引入β位取代基的绝对构型对化合物的活性有重要的影响, 一般一个构型的化合物活性明显高于其异构体.连接链一般为2~4个原子单元, 以醚键连接居多.芳香片段一般为多取代的联苯或取代芳香环结构; 对于芳香片段为联芳基的分子, 最外侧的苯环单元可以引入合适的亲水基团, 以改善化合物的理化性质.
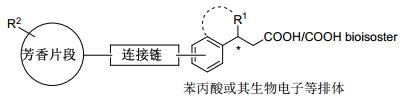 图 2
GPR40激动剂的药效团模型
Figure 2.
Pharmacophore model of GPR40 agonists
图 2
GPR40激动剂的药效团模型
Figure 2.
Pharmacophore model of GPR40 agonists
由于GPR40激动剂的药效团模型明确, 芳基丙酸为必需官能团, 构效关系及结构优化研究主要围绕图 2所示的三个组成模块进行, 研究思路比较相似.因此, 我们重点以已进入临床实验或临床前研究的候选药物分子为代表, 总结了有关GPR40激动剂研发的文章和专利, 分别针对三个模块的结构优化研究进行分类介绍, 综述该领域的最新研究进展.
1 Takeda公司TAK-875的研发历程
目前已有多家公司开发的GPR40激动剂进行过临床实验研究, 例如Takeda公司的TAK-875曾进入三期临床; Tobacco、Amgen、Eli Lilly、恒瑞和Piramal等公司的化合物也进行了二期或一期临床实验, 从而为GPR40作为抗糖尿病药靶的有效性提供了临床证据.其中, Takeda公司研发的TAK-875是临床研究进展最快的GPR40激动剂, 曾经进入三期临床(图 3). TAK-875的研发历程也极具代表性, 为GPR40激动剂的设计奠定了重要的基础, 故而我们首先介绍其研发历程, 并以此为模板介绍其他类型小分子GPR40激动剂的改造思路和结果.
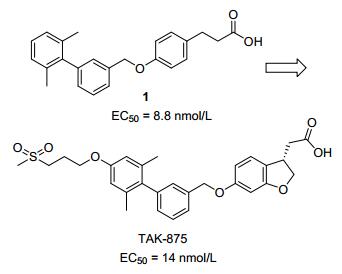 图 3
Takeda公司的GPR40激动剂
Figure 3.
GPR40 agonists developed by Takeda
图 3
Takeda公司的GPR40激动剂
Figure 3.
GPR40 agonists developed by Takeda
最初, Takeda公司的研究人员考虑到GPR40的内源性激动剂为中、长链脂肪酸, 所以他们对一系列含芳香环的脂肪酸进行了筛选, 结果发现苯丙酸在100 μmol/L下对GPR40有中等的激动活性.以此为基础, 经过深入的构效关系研究和结构改造, 如苯环取代基种类及其取代位置等, Takeda公司得到了先导化合物1(图 3).其中羧基远端苯环上的两个甲基可以使联苯结构中的两个苯环保持一定的二面角, 对提高活性十分必要.化合物1虽然具备了很高的体外活性(EC50=8.8 nmol/L), 但是其生物利用度仅为0.9%.研究人员推测其可能原因为羧基的β位易发生β氧化, 导致苯丙酸衍生物被快速代谢成苯甲酸衍生物[7].基于此设想, 研究人员通过在羧基β位引入合适取代基的方法, 有效地降低了β氧化, 提高了生物利用度[8].并且, 研究表明该取代基的绝对构型对活性有重要的影响, (S)-构型的二氢苯并呋喃衍生物活性明显高于(R)-构型对映体.进一步在2, 6-二甲基苯基上引入含砜基的亲水侧链, 调整化合物的理化性质, 得到了候选药物TAK-875. TAK-875的EC50=14 nmol/L, 其在肥胖Wistar大鼠的oGTT (口服葡萄糖耐量实验)模型上显示了剂量依赖的降血糖活性(0.3~3 mg/kg). TAK-875在大鼠和犬模型上, 口服给药后的半衰期分别为4.1和7.5 h, 生物利用度分别为76.0%和92.4%.其对相关GPCRs如GPR41、GPR43和GPR120等具有良好的选择性(EC50均大于10 μmol/L)[9].在进一步的临床实验中, TAK-875的半衰期显示了明显的种属差异, 其在人体的半衰期达到了28~30 h[10].当采用50 mg给药剂量, 每天给药一次时, 病人耐受性良好, 仅出现鼻咽炎或上呼吸道炎症副作用病例.并且其引起低血糖的风险明显低于对照组的磺酰脲类药物, 验证了TAK-875诱导的胰岛素分泌是血糖依赖的.虽然TAK-875表现了良好的疗效, 但是由于肝毒性问题, Takeda公司于2013年12月宣布终止其临床实验[11].
虽然TAK-875最终未能上市, 但是其研发思路及临床数据为GPR40激动剂的研究与开发提供了很好的参考. TAK-875与GPR40的晶体复合物结构于2014年被解析(图 4)[12], 这也为后续的合理药物设计提供了更多信息.晶体复合物结构显示, TAK-875的羧基与Arg183、Arg 258、Tyr 91和Tyr 240之间形成离子/氢键相互作用; Trp 174与二氢苯并呋喃平面呈垂直状; 2, 6-二甲基苯基4-位连接的亲水链伸向受体外, 故该位置可容忍较大改造.
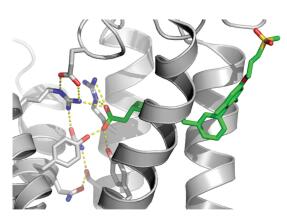 图 4
TAK-875与hGPR40的晶体复合物
Figure 4.
Co-crystal structure of hGPR40 and TAK-875
图 4
TAK-875与hGPR40的晶体复合物
Figure 4.
Co-crystal structure of hGPR40 and TAK-875
2 针对不同药效团部分的结构改造
目前文献报道的大部分GPR40激动剂在结构上均与TAK-875有相似之处.为了更清楚地介绍这些化合物的特点和研发思路, 我们以TAK-875为参照物, 依照药效团模型中的三个组成部分, 即针对丙酸侧链、连接链及芳香片段的结构改造, 分别介绍不同公司和机构研发的小分子GPR40激动剂.值得一提的是, 这三个方面的结构改造是交叉进行的, 只是不同化合物的侧重点或突破点在某一个模块的改造上, 因此被归入相应的结构改造类型.
2.1 侧重丙酸侧链的结构改造
2.2 侧重连接链的结构改造
2.3 侧重芳香片段的结构改造
2.1.3 Bristol-Myers Squibb公司
Bristol-Myers Squibb公司的专利陆续公布了一系列非苯丙酸类GPR40激动剂.该类分子中, 羧基不再是通过两个碳原子与苯环相连, 而是引入四氢吡咯[20]或二氢吡唑[21]作为连接板块, 即通过杂环将丙基链进行构像约束(图 7).该类GPR40激动剂同样表现了良好的活性, 如化合物2, 体外活性为EC50=6 nmol/L[22].
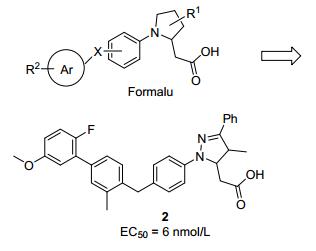 图 7
Bristol-Myers Squibb公司的GPR40激动剂
Figure 7.
GPR40 agonists developed by Bristol-Myers Squibb
图 7
Bristol-Myers Squibb公司的GPR40激动剂
Figure 7.
GPR40 agonists developed by Bristol-Myers Squibb
2.1.2 Piramal公司
2013年7月, Piramal公司的化合物P11187开始进行一期临床实验, 但是其结构也没有公布[18].该公司公布的专利保护了一系列丙酸侧链羧基β位引入氧杂环丁烷取代的化合物, 连接链及芳香片段与TAK-875类似(图 6).其中部分化合物EC50小于100 nmol/L[19].
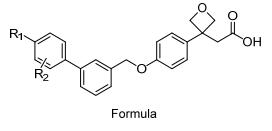 图 6
Piramal公司的GPR40激动剂
Figure 6.
GPR40 agonists developed by Piramal
图 6
Piramal公司的GPR40激动剂
Figure 6.
GPR40 agonists developed by Piramal
2.1.1 Amgen公司
Amgen公司是最早开展GPR40激动剂研发的制药公司之一.其研发的化合物AMG 837也是第一个进入临床实验的GPR40激动剂(图 5). AMG 837是GPR40部分激动剂, 其EC50=60 nmol/L.药物代谢实验表明, AMG 837在包括小鼠、大鼠、犬及猴等多个种属上均显示了良好的生物利用度.小鼠的oGTT表明, AMG837在10 mg/kg剂量下, 对于野生型小鼠表现出了显著的降血糖活性; 而对于GPR40敲除的小鼠, 并没有表现出明显的降血糖效果[13].良好的活性及药代性质, 使化合物AMG 837通过审批进入了一期临床实验.临床数据证实了预期的设想, 对于健康志愿者而言, AMG 837并不引起胰岛素水平升高及血糖降低[14].但是一期临床实验结束后, 关于AMG 837并没有后续报道, 可能由于其安全性仍然存在问题.其中一个重要因素在于AMG 837的亲脂性过高、极性表面小, 可能会进入中枢系统, 引起副作用[15].
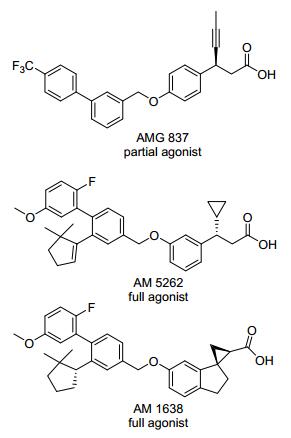 图 5
Amgen公司的GPR40激动剂
Figure 5.
GPR40 agonists developed by Amgen
图 5
Amgen公司的GPR40激动剂
Figure 5.
GPR40 agonists developed by Amgen
从2012年开始, Amgen公司又陆续报道了GPR40的完全激动剂AM 1638和AM 5262(图 5).该类完全激动剂不仅内在活性大大高于AMG 837, 而且能够激活Gs/cAMP通路, 促进GLP-1和GIP的释放, 是一类新机制的GPR40激动剂. AM 1638的主要结构特点是将AMG 837中处于对位的连接链和丙酸侧链改变成间位, 并且羧基β位的取代基由(S)-构型的丙炔基改变为(S)-构型的环丙基.与部分激动剂AMG 837相比, AM 1638虽然体外活性较低(EC50=160 nmol/L), 但是在BDF/DIO小鼠oGTT模型上, 60 mg/kg剂量下, AM 1638表现出了更高的促胰岛素分泌和降血糖活性[16].这提示了完全激动剂较部分激动剂可能会有更加优越的体内效果.对AM 1638的丙酸侧链进行构象约束得到了化合物AM 5262, 活性进一步提高至EC50=81 nmol/L.构象约束后使得分子脱靶效应明显降低, 实验表明AM 5262对于GPCRs、离子通道、转运体和酶的选择性优于AM 1638.另外AM 5262的清除率较AM 1638也有所下降, 体内活性明显优于AM 1638.在oGTT模型中, AM 5262在30 mg/kg剂量下显示出与AM 1638在60 mg/kg剂量下相当的降血糖活性[17].
2.2.1 恒瑞公司
恒瑞公司研发的化合物呋格列泛目前正处于一期临床实验.呋格列泛的连接链采用构象约束策略, 形成二氧六环与苯环并环的结构(图 8).呋格列泛的开发源于对TAK-875的改造.起初, 研究人员直接在TAK-875分子的基础上引入一个并环, 合成了化合物3(如图 8), 结果显示活性得到了保持.接着研究人员发现, 引入二氧六环并环后, 删除2, 6-二甲基苯基片段, 结构简化的构像约束化合物仍可保持良好的活性, 有效降低了分子量.体内实验显示, 在猴的ivGTT (静脉葡萄糖耐量实验)模型中, 给药剂量为6 mg/kg, 呋格列泛降血糖活性优于TAK-875[23].但是呋格列泛在小鼠模型上的降血糖效果不佳, 即使在50 mg/kg剂量下, 降低血糖量也仅为4.9%.这一结果有可能是由于GPR40的种属差异造成的.呋格列泛在猴模型上降血糖活性得到确证, 支持了其进入接下来的临床实验.
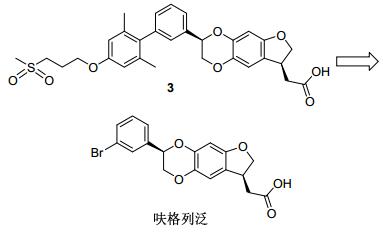 图 8
恒瑞公司的GPR40激动剂
Figure 8.
GPR40 agonists developed by Hengrui
图 8
恒瑞公司的GPR40激动剂
Figure 8.
GPR40 agonists developed by Hengrui
2.2.4 Sanofi-Aventis公司
Sanofi-Aventis公司早期报道了长链脂肪酸衍生物类GPR40激动剂, 但是活性并不理想, 如化合物6的EC50=740 nmol/L(图 11)[29].接下来, 该公司研究人员针对具有同等活性的构象约束化合物7(EC50=630 nmol/L), 将其连接链噁唑环开环[30], 以乙/丙二醇[31]或草酰胺[32]作为连接链时, 化合物活性明显提高(图 11). SAR1的体外活性达到EC50=0.4 nmol/L.在肥胖ZDF大鼠oGTT模型上, SAR1的最小有效剂量为1 mg/kg[33].
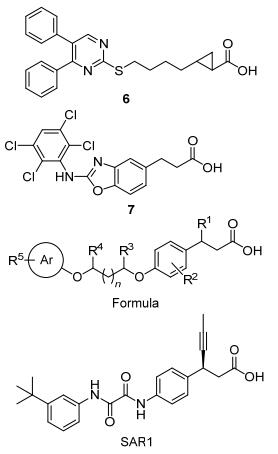 图 11
Sanofi-Aventis公司的GPR40激动剂
Figure 11.
GPR40 agonists developed by Sanofi-Aventis
图 11
Sanofi-Aventis公司的GPR40激动剂
Figure 11.
GPR40 agonists developed by Sanofi-Aventis
2.2.2 南丹麦大学(University of Southern Denmark)
Ulven Trond课题组长期致力于GPR40激动剂的研究, 其开发的一系列GPR40激动剂的结构特点在于连接链的改变.与常见的醚类连接方式不同, 该类GPR40激动剂的连接链为刚性的碳-碳三键. Ulven Trond等考虑到很多大于10个碳的饱和或不饱和脂肪酸均会对GPR40产生一定的激动活性, 所以他们尝试筛选了一系列结构狭长的亲脂性羧酸, 结果成功地得到了活性高于油酸10倍的化合物4(图 9).接着他们以此为苗子化合物, 通过构效关系研究和结构优化, 得到了先导化合物TUG-424 (EC50=46 nmol/L), 其在100 nmol/L浓度下可以促进大鼠胰岛素分泌细胞系INS-1E的糖促胰岛素分泌(图 9)[24].接下来该课题组对先导化合物TUG-424进行进一步成药性优化, 主要集中在降低分子亲脂性和提高代谢稳定性[25].化合物TUG-424的LogD=2.4, 在其芳香片段2-位的甲基上引入氰基后, 得到化合物TUG-488, 活性提高至EC50=20 nmol/L, 同时LogD下降至1.3(如图 9), 药代性质有所改善, 血浆暴露量明显提高, 约为TUG-424的3.5倍.在小鼠oGTT模型中, TUG-488在10 mg/kg剂量下显示了与DPP4抑制剂sitagliptin类似的降血糖活性.但是TUG-488的半衰期仍然很短, 仅有50 min[25b].为了进一步延长该类GPR40激动剂的半衰期, 研究人员在丙酸侧链的邻位引入了氟原子, 得到化合物TUG-770 (如图 9).与TUG-488相比, TUC-770活性提高至EC50=6 nmol/L, 血浆暴露量显著提升, 而且半衰期也由50 min提高至355 min.在大鼠ipGTT(腹腔葡萄糖耐量实验)模型中, TUG-770在50 mg/kg给药剂量下降血糖活性明显高于sitagliptin (10 mg/kg)[26].
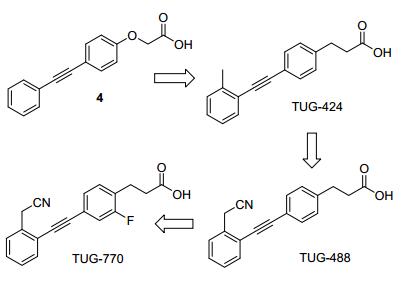 图 9
Ulven Trond课题组的GPR40激动剂
Figure 9.
GPR40 agonists developed by Ulven Trond's group
图 9
Ulven Trond课题组的GPR40激动剂
Figure 9.
GPR40 agonists developed by Ulven Trond's group
2.2.3 Daiichi Sankyo公司
Daiichi Sankyo公司报道的GPR40激动剂DS-1558, 不仅具有高活性(EC50=3.7 nmol/L), 而且分子量相对较小(图 10), 其连接链与芳基部分进行了成环修饰.研究人员发现, 在羧基β位引入乙氧基后, 可以将芳香片段简化为单取代苯环, 如外消旋体5(EC50=20 nmol/L)[27].但是5的口服半衰期较短, 仅有0.37 h.通过对代谢产物的分析, 研究人员认为芳香片段中的甲基是该化合物的主要代谢位点.故而他们将芳香片段与连接链环在一起以封闭该代谢位点, 得到化合物DS-1558.该改造不仅提高了体外活性, 而且药代性质也明显改善, 半衰期和血浆暴露量较5均有提高. DS-1558在多个种属如大鼠、犬和猴上均显示了良好的生物利用度.在ZDF大鼠oGTT模型上, DS-1558在0.1 mg/kg剂量即达到sitagliptin在10 mg/kg剂量下的同等降血糖水平.[28]值得一提的是, 该公司的研究人员认为, GPR40激动剂通常具有的联苯结构会抑制线粒体呼吸链中的complex1, 由此带来细胞毒的风险[27].因此, 他们通过在羧基β位引入乙氧基从而去除联苯结构(5, 图 10), 也被认为是该类分子的一大优势.
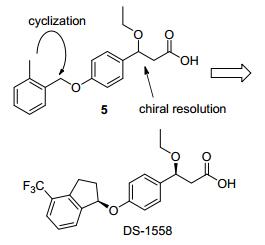 图 10
Daiichi Sankyo公司的GPR40激动剂
Figure 10.
GPR40 agonists developed by Daiichi Sankyo
图 10
Daiichi Sankyo公司的GPR40激动剂
Figure 10.
GPR40 agonists developed by Daiichi Sankyo
2.2.5 Merck公司
Merck公司先后开发了多种结构类型的GPR40激动剂.其早期研发的GPR40激动剂主要是二芳基醚类衍生物, 其中又分为两类:经典的苯丙酸类衍生物和可作为羧基电子等排体的噻唑烷二酮类衍生物(图 12).噻唑烷二酮类衍生物8的体外活性为EC50=10 nmol/L; 在野生型小鼠的ipGTT模型上, 给药剂量为30 mg/kg时, 可观察到降血糖活性; 对于GPR40敲除的小鼠, 同等给药剂量下, 化合物8未表现出明显的降血糖活性[34].苯丙酸类衍生物9的体外活性EC50=71 nmol/L; 在小鼠的ipGTT模型上, 给药剂量为10 mg/kg时, 可观察到降血糖活性.化合物9具有很高的生物利用度, 半衰期为7.8 h[35].
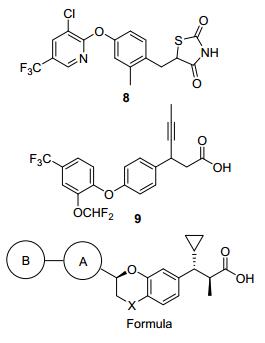 图 12
Merck公司的GPR40激动剂
Figure 12.
GPR40 agonists developed by Merck
图 12
Merck公司的GPR40激动剂
Figure 12.
GPR40 agonists developed by Merck
2014年Merck公司公布的专利中报道了一系列新型的高活性GPR40激动剂.研究人员在羧基的α和β位分别引入手性的甲基和环丙基, 并且将连接链与苯丙酸片段中的苯环成环, 以得到构象更加固定的分子(图 12).这一改造极大的提高了化合物的活性, 其中部分化合物的EC50<1 nmol/L[36].
2.3.1 Tobacco公司
Tobacco公司的化合物JTT-851曾经进入二期临床实验, 但是其具体结构至今尚未公布. Tobacco公司公布的专利报道了一系列螺环类GPR40激动剂(图 13).该类分子中的醚类连接链所连接的不是芳香环, 而是含双键的螺环结构[37].
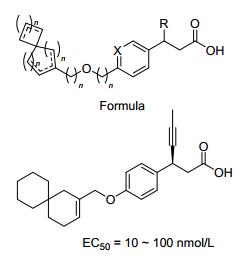 图 13
Tobacco公司的GPR40激动剂
Figure 13.
GPR40 agonists developed by Tobacco
图 13
Tobacco公司的GPR40激动剂
Figure 13.
GPR40 agonists developed by Tobacco
2.3.2 Eli Lilly公司
Eli Lilly公司曾经进入一期临床的化合物LY2881835, 芳香片段不再是两个苯环直接相连, 而是由哌啶环连接的两个芳香体系(图 14)[38]. LY2881835体外对GPR40的激动活性为EC50=233 nmol/L.体内实验显示, 在ipGTT模型上其EC50=0.14 mg/kg; 在大鼠的oGTT模型上, LY2881835显示了优于罗格列酮的剂量依赖的降血糖活性. 2011年8月, Eli Lilly公司在美国完成了LY2881835的一期临床实验, 但是由于副作用原因, 该化合物并未被继续推进[39].
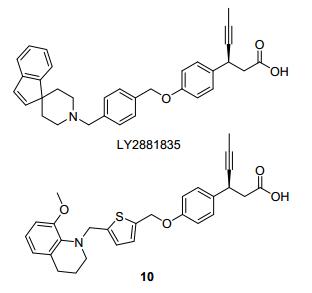 图 14
Eli Lilly公司的GPR40激动剂
Figure 14.
GPR40 agonists developed by Eli Lilly
图 14
Eli Lilly公司的GPR40激动剂
Figure 14.
GPR40 agonists developed by Eli Lilly
2013年, Eli Lilly公司公开的专利中报道了LY2881835的类似物10(图 14), 其体外EC50=150 nmol/L.在MIN6细胞水平上表现出剂量依赖的促胰岛素分泌活性(1~10 μmol/L); 体内ipGTT的ED50=1 mg/kg[40].
2.3.3 Connexios公司
Connexios公司的专利中保护了一类新结构GPR40激动剂, 其中部分化合物EC50<10 nmol/L.该类分子的芳香片段部分含有亚胺结构, 多数活性分子的羧基β位为氰基取代, 但是该公司并未公布具体哪个构型活性更高(图 15)[41].该公司在2013年报道了其研发的高活性GPR40激动剂CNX-011-67 (EC50=0.24 nmol/L), 以5 mg/kg给药剂量, 在ZDF大鼠上为期7周的实验[42].结果显示, CNX-011-67不仅能够诱导葡萄糖依赖的胰岛素分泌, 降低血清中游离脂肪酸和甘油三酯含量, 还能够减少胰岛β细胞的凋亡.但是CNX-011-67的具体结构尚未公布.
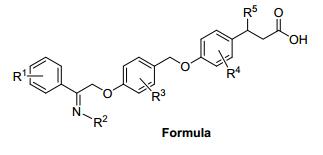 图 15
Connexios公司的GPR40激动剂
Figure 15.
GPR40 agonists developed by Connexios
图 15
Connexios公司的GPR40激动剂
Figure 15.
GPR40 agonists developed by Connexios
3 总结与展望
GPR40作为当前抗Ⅱ型糖尿病研究的热门靶点, 吸引了全球大量制药公司和科研机构的关注, 一系列高活性的GPR40激动剂相继被报道.但是, 由于Takeda公司的TAK-875因肝毒性问题终止于三期临床研究, 所以科研人员对该靶点的态度也开始转向谨慎.不过, 因为GPR40在肝脏并没有分布, 所以TAK-875导致的肝毒性可能与靶点无关; 而且, TAK-875的临床数据也显示GPR40激动剂具有降低Ⅱ型糖尿病病人HbA1c及空腹血糖的良好疗效.因此, 以GPR40作为抗Ⅱ型糖尿病靶点的药物研发并未因之而停止.随着研究的深入, 接下来在GPR40激动剂的开发过程中, 更多问题可能需要被重点关注, 比如化合物脂溶性、激动剂类型、亚型选择性等.这也使得安全有效的新型GPR40激动剂的研发变得更具挑战性.随着最近新结构GPR40激动剂被不断报道, 也为今后GPR40激动剂的研究与开发奠定了良好的基础.安全性更高的新型GPR40激动剂也很可能在未来的抗Ⅱ型糖尿病领域中扮演重要的角色.
-
- [1]
-
[2]
Ahren, B. Nat. Rev. Drug Discovery 2009, 8, 369. doi: 10.1038/nrd2782
-
[3]
Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; Tanaka, H.; Maruyama, M.; Satoh, R.; Okubo, S.; Kizawa, H.; Komatsu, H.; Matsumura, F.; Noguchi, Y.; Shinobara, T.; Hinuma, S.; Fujisawa, Y.; Fujino, M. Nature 2003, 422, 173. doi: 10.1038/nature01478
-
[4]
Edfalk, S.; Steneberg, P.; Edlund, H. Diabetes 2008, 57, 2280. doi: 10.2337/db08-0307
-
[5]
Nakamoto, K.; Nishinaka, T.; Matsumoto, K.; Kasuya, F.; Mankura, M.; Koyama, Y.; Tokuyama, S. Brain Res. 2012, 1432, 74. doi: 10.1016/j.brainres.2011.11.012
-
[6]
Hauge, M.; Vestmar, M. A.; Husted, A. S.; Ekberg, J. P.; Wright, M. J.; Di Salvo, J.; Weinglass, A. B.; Engelstoft, M. S.; Madsen, A. N.; Luckmann, M.; Miller, M. W.; Trujillo, M. E.; Frimurer, T. M.; Holst, B.; Howard, A. D.; Schwartz, T. W. Mol. Metabolism 2015, 4, 3. doi: 10.1016/j.molmet.2014.10.002
-
[7]
Sasaki, S.; Kitamura, S.; Negoro, N.; Suzuki, M.; Tsujihata, Y.; Suzuki, N.; Santou, T.; Kanzaki, N.; Harada, M.; Tanaka, Y.; Kobayashi, M.; Tada, N.; Funami, M.; Tanaka, T.; Yamamoto, Y.; Fukatsu, K.; Yasuma, T.; Momose, Y. J. Med. Chem. 2011, 54, 1365. doi: 10.1021/jm101405t
-
[8]
Negoro, N.; Sasaki, S.; Ito, M.; Kitamura, S.; Tsujihata, Y.; Ito, R.; Suzuki, M.; Takeuchi, K.; Suzuki, N.; Miyazaki, J.; Santou, T.; Odani, T.; Kanzaki, N.; Funami, M.; Tanaka, T.; Yasuma, T.; Momose, Y. J. Med. Chem. 2012, 55, 1538. doi: 10.1021/jm2012968
-
[9]
Negoro, N.; Sasaki, S.; Mikami, S.; Ito, M.; Suzuki, M.; Tsujihata, Y.; Ito, R.; Harada, A.; Takeuchi, K.; Suzuki, N.; Miyazaki, J.; Santou, T.; Odani, T.; Kanzaki, N.; Funami, M.; Tanaka, T.; Kogame, A.; Matsunaga, S.; Yasuma, T.; Momose, Y. ACS Med Chem. Lett. 2010, 1, 290. doi: 10.1021/ml1000855
-
[10]
Burant, C. F. Diabetes Care 2013, 36, 175.
- [11]
-
[12]
Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J.; Jennings, A.; Okada, K. Nature 2014, 513, 124. doi: 10.1038/nature13494
-
[13]
Houze, J. B.; Zhu, L. S.; Sun, Y.; Akerman, M.; Qiu, W.; Zhang, A. J.; Sharma, R.; Schmitt, M.; Wang, Y. C.; Liu, J. W.; Liu, J. I. A.; Medina, J. C.; Reagan, J. D.; Luo, J.; Tonn, G.; Zhang, J.; Lu, J. Y. L.; Chen, M.; Lopez, E.; Nguyen, K.; Yang, L.; Tang, L.; Tian, H.; Shuttleworth, S. J.; Lin, D. C. H. Bioorg. Med. Chem. Lett. 2012, 22, 1267. doi: 10.1016/j.bmcl.2011.10.118
-
[14]
Luo, J.; Swaminath, G.; Brown, S. P.; Zhang, J.; Guo, Q.; Chen, M.; Nguyen, K.; Tran, T.; Miao, L.; Dransfield, P. J.; Vimolratana, M.; Houze, J. B.; Wong, S.; Toteva, M.; Shan, B.; Li, F.; Zhuang, R.; Lin, D. C. H. PLOS One 2012, 7, 46300. doi: 10.1371/journal.pone.0046300
-
[15]
Liu, J. W.; Wang, Y. C.; Ma, Z. H.; Schmitt, M.; Zhu, L. S.; Brown, S. P.; Dransfield, P. J.; Sun, Y.; Sharma, R.; Guo, Q.; Zhuang, R.; Zhang, J.; Luo, J.; Tonn, G. R.; Wong, S.; Swaminath, G.; Medina, J. C.; Lin, D. C. H.; Houze, J. B. ACS Med. Chem. Lett. 2014, 5, 517. doi: 10.1021/ml400501x
-
[16]
Brown, S. P.; Dransfield, P. J.; Vimolratana, M.; Jiao, X. Y.; Zhu, L.; Pattaropong, V.; Sun, Y.; Liu, J. Q.; Luo, J.; Zhang, J.; Wong, S.; Zhuang, R.; Guo, Q.; Li, F.; Medina, J. C.; Swaminath, G.; Lin, D. C. H.; Houze, J. B. ACS Med. Chem. Lett. 2012, 3, 726. doi: 10.1021/ml300133f
-
[17]
Wang, Y. C.; Liu, J. W.; Dransfield, P. J.; Zhu, L. S.; Wang, Z. Y.; Du, X. H.; Jiao, X. Y.; Su, Y. L.; Li, A. R.; Brown, S. P.; Kasparian, A.; Vimolratana, M.; Yu, M.; Pattaropong, V.; Houze, J. B.; Swaminath, G.; Tran, T.; Nguyen, K.; Guo, Q.; Zhang, J.; Zhuang, R.; Li, F.; Miao, L.; Bartberger, M. D.; Correll, T. L.; Chow, D.; Wong, S.; Luo, J.; Lin, D. C. H.; Medina, J. C. ACS Med. Chem. Lett. 2013, 4, 551. doi: 10.1021/ml300427u
- [18]
-
[19]
Kumar, S.; Sharma, R.; Mahajan, V. A.; Sawargave, S. P. WO 2013128378, 2013 [Chem. Abstr. 2013, 159, 455418].
-
[20]
Ellsworth, B. A.; Ewing, W. R.; Jurica, E. US 0082165, 2011 [Chem. Abstr. 2011, 154, 434695].
-
[21]
Hernandez, A. S.; Ellsworth, B. A.; Ewing, W. R.; Chen, B. WO 2014078608, 2014 [Chem. Abstr. 2014, 160, 740116].
-
[22]
Abdel-Magid, A. F. ACS Med. AChem. ALett. 2014, 5, 954. doi: 10.1021/ml5002757
-
[23]
Lu, H. J.; Fei, H. B.; Yang, F. L.; Zheng, S. X.; Hu, Q. Y.; Zhang, L.; Yuan, J. J.; Feng, J.; Sun, P. Y.; Dong, Q. Bioorg. AMed. AChem. ALett. 2013, 23, 2920. doi: 10.1016/j.bmcl.2013.03.060
-
[24]
Christiansen, E.; Urban, C.; Merten, N.; Liebscher, K.; Karlsen, K. K.; Hamacher, A.; Spinrath, A.; Bond, A. D.; Drewke, C.; Ullrich, S.; Kassack, M. U.; Kostenis, E.; Ulven, T. J. Med. Chem. 2008, 51, 7061. doi: 10.1021/jm8010178
-
[25]
Christiansen, E.; Urban, C.; Grundmann, M.; Due-Hansen, M. E.; Hagesaether, E.; Schmidt, J.; Pardo, L.; Ullrich, S.; Kostenis, E.; Kassack, M.; Ulven, T. J. Med. Chem. 2011, 54, 6691.
(b) Christiansen, E; Due-Hansen, M; E; Urban, C; Grundmann, M; Schmidt, J; Hansen, S; V; F; Hudson, B; D; Zaibi, M; Markussen, S; B; Hagesaether, E; Milligan, G; Cawthorne, M; A; Kostenis, E; Kassack, M; U; Ulven, T; J. Med. Chem. 2013, 56, 982; -
[26]
Christiansen, E.; Hansen, S. V. F.; Urban, C.; Hudson, B. D.; Wargent, E. T.; Grundmann, M.; Jenkins, L.; Zaibi, M.; Stocker, C. J.; Ullrich, S.; Kostenis, E.; Kassack, M. U.; Milligan, G.; Cawthorne, M. A.; Ulven, T. ACS Med. Chem. Lett. 2013, 4, 441. doi: 10.1021/ml4000673
-
[27]
Takano, R.; Yoshida, M.; Inoue, M.; Honda, T.; Nakashima, R.; Matsumoto, K.; Yano, T.; Ogata, T.; Watanabe, N.; Toda, N. Bioorg. Med. Chem . Lett. 2014, 24, 2949. doi: 10.1016/j.bmcl.2014.04.065
-
[28]
Takano, R.; Yoshida, M.; Inoue, M.; Honda, T.; Nakashima, R.; Matsumoto, K.; Yano, T.; Ogata, T.; Watanabe, N.; Hirouchi, M.; Yoneyama, T.; Ito, S.; Toda, N. ACS Med. Chem. Lett. 2015, 6, 266. doi: 10.1021/ml500391n
-
[29]
Defossa, E.; Goerlitzer, J.; Klabunde, T.; Drosou, V.; Stengelin, S.; Haschke, G.; Herling, A.; Bartoschek, S. WO 2007131619, 2007 [Chem. Abstr. 2007, 148, 11254].
-
[30]
Defossa, E.; Follmann, M.; Klabunde, T.; Drosou, V.; Hessler, G.; Stengelin, S.; Haschke, G.; Herling, A.; Bartoschek, S. WO 2007131622, 2007 [Chem. Abstr. 2007, 147, 541859].
-
[31]
Kell, S.; Deffossa, E.; Dietrich, V.; Stengelin, S.; Herling, A.; Haschke, G.; Klabunde, T. US 20120004166, 2012 [Chem. Abstr. 2012, 156, 122146].
(b) Kell, S.; Deffossa, E.; Dietrich, V.; Stengelin, S.; Herling, A.; Haschke, G.; Klabunde, T. US 20120004187, 2012 [Chem. Abstr. 2012, 156, 122147]. -
[32]
Deffossa, E.; Dietrich, V.; Klabunde, T.; Kell, S.; Stengelin, S.; Haschke, G.; Herling, A.; Kuhlmann-Gottke, J.; Bartoschek, S.; Gessler, S.; Dudda, A.; Billen, G.; Ollp, T.; Rieke-Zapp, J. US 20130172248, 2013 [Chem. Abstr. 2013, 159, 181654 ].
-
[33]
Defossa, E.; Wagner, M. Bioorg. Med. Chem. Lett. 2014, 24, 2991. doi: 10.1016/j.bmcl.2014.05.019
-
[34]
Zhou, C. Y.; Tang, C.; Chang, E.; Ge, M.; Lin, S. N.; Cline, E.; Tan, C. P.; Feng, Y.; Zhou, Y. P.; Eiermann, G. J.; Petrov, A.; Salituro, G.; Meinke, P.; Mosley, R.; Akiyama, T. E.; Einstein, M.; Kumar, S.; Berger, J.; Howard, A. D.; Thornberry, N.; Mills, S. G.; Yang, L. H. Bioorg. Med.Chem. Lett. 2010, 20, 1298. doi: 10.1016/j.bmcl.2009.10.052
-
[35]
Walsh, S. P.; Severino, A.; Zhou, C. Y.; He, J. F.; Liang, G. B.; Tan, C. P.; Cao, J.; Eiermann, G. J.; Xu, L.; Salituro, G.; Howard, A. D.; Mills, S. G.; Yang, L. H. Bioorg. Med. Chem Lett. 2011, 21, 3390. doi: 10.1016/j.bmcl.2011.03.114
-
[36]
Brockunier, L. L.; Chen, H.; Chobanian, H. R.; Clements, M. J.; Crespo, A.; Demong, D. E.; Guo, Y.; Hagmann, W. K.; Marcantonio, K. M.; Miller, M.; Pio, B.; Plummer, C. W.; Xiao, D. WO 2014130608, 2014 [Chem. Abstr. 2014, 161, 419304].
-
[37]
Shimada, T.; Ueno, H.; Tsutsumi, K.; Aoyagi, K.; Manabe, T.; Sasaki, S.; Katoh, S. WO 2009054479, 2009 [Chem. Abstr. 2009, 150, 494618].
-
[38]
Hamdouchi, C.; Lineswala, J. P.; Maiti, P. WO 2011046851, 2011 [Chem. Abstr. 2011, 154, 486226].
- [39]
-
[40]
Hamdouchi, C. WO 2013025424, 2013 [Chem. Abstr. 2013, 158, 331040].
-
[41]
Rao, J. M. R.; Arumugam, N.; Ansari, M. M.; Gudla, C.; Pachiyappan, S.; Ramalingam, M.; George, J.; Arul, G. F.; Bommegowda, Y. K.; Angupillai, S. K.; Kottamalai, R.; Jidugu, P.; Rao, D. S. WO 2012011125, 2012 [Chem. Abstr. 2012, 156, 202874].
-
[42]
Gowda, N.; Dandu, A.; Singh, J.; Biswas, S.; Raghav, V.; Lakshmi, M. N.; Shilpa, P. C.; Sunil, V.; Reddy, A.; Sadasivuni, M.; Aparna, K.; Verma, M. K.; Moolemath, Y.; Anup, M. O.; Venkataranganna, M. V.; Somesh, B. P.; Jagannath, M. R. BMC Pharmacol. Toxicol. 2013, 14, 28. doi: 10.1186/2050-6511-14-28
-

图 7 Bristol-Myers Squibb公司的GPR40激动剂
Figure 7 GPR40 agonists developed by Bristol-Myers Squibb
-


 下载:
下载:














 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 991
- HTML全文浏览量: 273
 下载:
下载:
