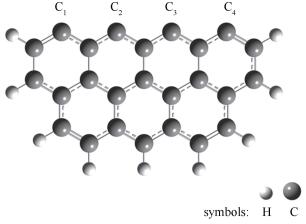
图 1.
Zigzag型焦炭模型示意图
Figure 1.
Char model with zigzag edge (carbon atoms at the edge were labeled as: C1, C2, C3, C4)

燃煤氮氧化物(NOx)排放是目前大气污染治理的重点。再燃(或称燃料分级)技术灵活可调、适应强,与低NOx燃烧器(LNB)、选择性非催化还原(SNCR)以及选择性催化还原(SCR)兼容性好,已被证实是一种发展潜力较大的炉内燃烧调整脱硝技术[1]。再燃燃料可选种很多,包括气体燃料(天然气等)、液体燃料(水煤浆等)及固体燃料(超细煤粉、农林废弃物、市政垃圾等)。其中,农林废弃物等生物质作为一种环境友好的可再生能源,是近年来再燃燃料研究的重点。通常意义上讲,生物质再燃还原NO包括挥发分的均相还原及焦炭的异相还原。目前,关于挥发分均相还原NO已有大量的报道[2-4],但焦炭异相还原NO过程较为复杂,现阶段还缺乏系统、深入的认识,成为限制生物质再燃脱硝进一步工程应用的重要阻碍因素。
另一方面,与燃煤相比,生物质燃料中的碱金属含量高,不仅会增加锅炉尾部烟道腐蚀的风险,而且势必会对其再燃脱硝产生相应的影响[5-7]。钠是生物质灰分中含量较高的一种碱金属元素,已有大量文献从实验和理论两方面研究了钠对焦炭异相还原NO的作用机理。刘银河等[8]发现,碱金属Na能够抑制异相反应过程中NO及其前驱物的形成,但并未指明具体的催化机理。Zhao等[9]发现,Na促进煤粉燃烧的同时,对NOx也具有高催化活性。Zhang等[10]通过密度泛函理论研究发现负载Na可以增强焦炭对NO分子的吸附能力,但缺乏后续的NO还原研究。温正城等[11]采用量子力学计算发现,Na的存在显著降低了煤焦异相还原NO的活化能,但计算只选用单个碳原子作为焦炭模型,且在计算Na的催化作用时未考虑焦炭的影响,未能准确体现Na对NO异相还原的作用。综上,目前,基于分子水平对Na影响焦炭异相还原NO的反应机理研究不足。
本研究基于密度泛函理论(DFT)和经典过渡态理论(TST),借助分子模拟的手段探究了生物质再燃过程中焦炭对NO的异相还原机理,并重点结合反应动力学计算,分析了生物质所含碱金属Na对焦炭异相还原NO的影响机制。本研究所得结论完善了生物质焦异相还原NO的基础理论,为再燃脱硝的工程应用提供了理论指导。
已有的研究表明,煤、生物质等含碳固体燃料高温热解产物主要是由3-7个苯环组成的大量芳香环簇结构不规则堆积而成[12],且边缘锯齿型(zigzag)及扶手型(armchair)苯环簇模型已被证实为模拟焦炭计算的理想模型[13, 14],但研究发现,由于zigzag型焦炭模型的边缘存在未成对电子,因此,其活性位点更为活跃[15, 16]。钟俊等[17]利用量子化学计算采用七环锯齿形苯环簇模型对煤焦异相还原N2O的反应机理进行了研究并取得了理想的结果。朱恒毅等[18]选用相同模型,采用密度泛理论在分子水平上研究了富氧燃烧条件下了CO对煤焦异相还原NO反应的影响。因此,本研究选取zigzag型焦炭模型进行模拟计算,优化后的具体模型见图 1,焦炭模型上部的不饱和边缘代表游离的活性位点,其余碳原子则添加氢原子饱和。
本研究基于经典过渡态理论(TST),采用密度泛函理论(DFT),设计了两条反应路径,分别为焦炭异相还原NO以及碱金属Na元素参与的焦炭异相还原NO,所有计算均利用Dmol3模块完成。电子的交换关联作用选取广义梯度近似(GGA),采用Becke-Lee-Yang-Parr(BLYP)相泛函算法。计算精度在进行反应物、过渡态、中间体、产物的几何结构优化时设置为Fine(energy: 2.6255×10-2kJ/mol; Max force:52.51kJ/(mol·nm);Max displacement: 5×10-4 nm),自洽场(SCF)的总能量收敛极限为1.0×10-6 Ha,在过渡态搜索(transition state,TS)时设置为Medium(RMS convergence: 262.55kJ/(mol·nm))。采用加极化函数展开的双数值基组(DNP)处理价电子波函数,核心电子使用DFT Semi-core Psuedopots处理,同时计算采用Tkatchenko-Scheffler方法进行DFT-D修正,所有计算均考虑自选非限制性(Spin:unrestricted),且将多重度设为自动(Multiplicity:Auto)。为加速收敛并保证计算结果的准确,热拖尾效应(smearing)设置为13.13kJ/mol。使用频率振动分析证明各优化结构的合理性,结果表明产物、中间体及产物无虚频,过渡态存在唯一虚频。同时,采用相同计算水平对优化后的结构进行单点能计算,并在此基础上进行零点能校正,本研究中所有计算均基于校正后的能量进行。
利用经典过渡态理论(TST)对反应的决速步进行反应动力学分析,反应速率常数计算如公式(1)[19]所示。
|
$ k = \varGamma \times \frac{{{k_{\text{B}}}T}}{h} \times \frac{{{Q_{{\text{TS}}}}}}{{{Q_{\text{A}}}{Q_{\text{B}}}}} \times {\text{exp}}\left( {\frac{{ - {E_{\text{a}}}}}{{RT}}} \right) $ |
(1) |
式中,Γ为量子隧道修正系数;kB为玻尔兹曼常数,J/K;h为普朗克常数,J·s;QTS、QA、QB依次为过渡态TS、反应物A和反应物B的配分函数;T为温度,K;Ea为反应活化能,kJ/mol;R为气体摩尔常数,J/(mol·K);其中,量子隧道修正系数经验表达式见式(2)。
|
$ \varGamma = 1 + \left( {\frac{1}{{24}}} \right) \times {\left( {\frac{{h{v_{\text{m}}}c}}{{{k_{\text{B}}}T}}} \right)^2} $ |
(2) |
式中,vm为反应路径振动频率,cm-1;c为光速,m/s。
对反应过程中所有反应物、中间体、生成物及过渡态进行结构优化和零点能校正后的能量(E)及过渡态的唯一振动虚频如表 1所示,经结构优化后的反应物R(焦炭及NO)、主要生成物P(N2)的几何参数见表 2。通过表 1中数据可知,焦炭C、NO及N2经优化后的几何参数与以往文献中的实验值吻合良好,证明了本研究计算所用模型的准确性。
 下载:
导出CSV
下载:
导出CSV
| Species | E/(kJ·mol-1) | Frequency/cm-1 | Species | E/(kJ·mol-1) | Frequency /cm-1 |
| IM | -2856326.30 | - | NaIM2 | -3624341.19 | - |
| IM1 | -3197558.53 | - | NaIM3 | -3624339.89 | - |
| IM2 | -3197908.82 | - | NaP+N2 | -3624293.46 | - |
| IM3 | -3197907.32 | - | NaTS1 | -3623920.36 | -263.48 |
| P+N2 | -3197839.39 | - | NaTS2 | -3624328.91 | -233.86 |
| TS1 | -3197483.39 | -475.11 | NaTS3 | -3624234.95 | -165.53 |
| TS2 | -3197895.36 | -264.72 | NO | -341168.95 | - |
| TS3 | -3197768.49 | -131.19 | N2 | -287649.68 | - |
| NaIM | -3282727.29 | - | R | -2514653.09 | - |
| NaIM1 | -3623928.51 | - | Na | -426013.72 | - |
下载:
导出CSV
已有的研究表明,NO分子倾向于吸附在焦炭边缘且第一个NO分子最常以side-on模式与C2、C3活性位点相结合的方式吸附[23],而第二个NO分子以N-N结合方式及O-down的模式吸附时更利于N2的产生[24],在此基础上,本研究优化得到的反应物、过渡态、中间体及产物的几何构型、主要的键长及过渡态虚频如图 2所示,反应过程的反应势能面见图 3。
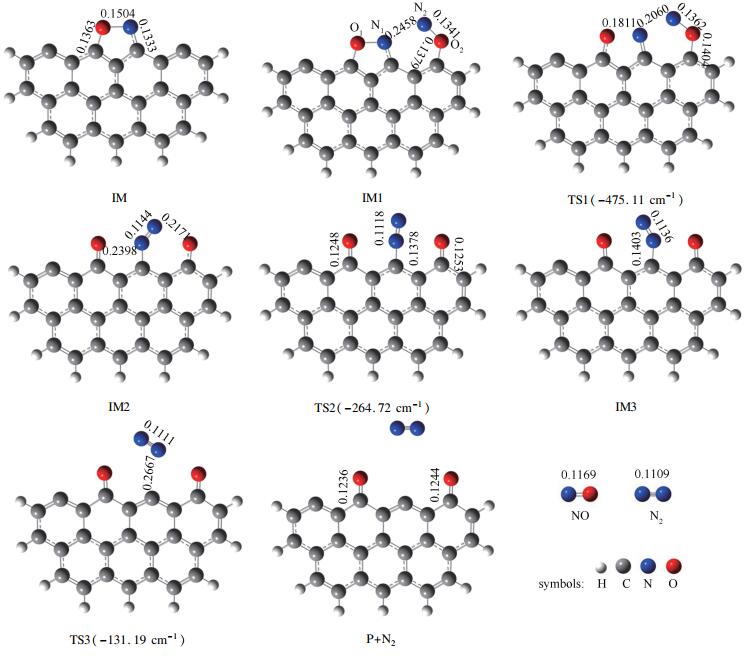
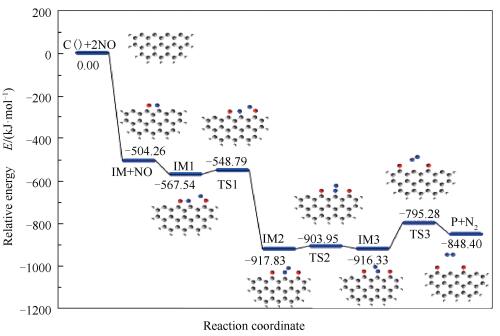
由图 2和图 3可知,该反应路径需要三个相邻的碳活性位。首先一个NO分子以N-O键轴平行于焦炭边缘的side-on模式与相邻的两个碳活性位点C2、C3相结合,形成具有五元环结构的稳定中间体IM。NO气体分子的N1-O1键长由自由状态下的0.1169nm伸长至0.1504nm,说明在NO分子吸附后N1-O1键趋于分离,同时释放504.26kJ/mol热量。然后第二个NO分子以O-down模式吸附于碳活性位点C4生成O2-C4键并形成吸附产物IM1,N2-O2键被由初始的0.1169nm拉长至0.1341nm,证明第二个NO分子吸附于焦炭边缘后亦倾向于分离,该过程释放63.28kJ/mol热量。随后,N2逐渐远离O2并向N1靠近,N2-O2键长由0.1341nm(IM1)→0.1362nm(TS1)→0.2171nm(IM2),而N1-N2键长则由0.2458nm(IM1)→0.2060nm(TS1)→0.1144nm(IM2),说明在该过程中N2-O2键逐渐减弱而N1-N2键显著增强,最后导致N2-O2键及断裂而形成N1-N2键,完成由IM1至IM2转变并克服18.75kJ/mol能垒,同时释放350.29kJ/mol能量。N1-N2键绕C3旋转形成了IM3,随后N1-C3键由0.1403nm(IM3)→0.2667nm(TS3)→∞(P),N1-C3键断裂且N2由焦炭边缘解吸附。此过程翻越121.04kJ/mol能垒,吸收67.92kJ/mol能量。由反应过程可知,IM3→P+N2为焦炭异相还原NO决速步,所需活化能为121.04kJ/mol。该反应过程C+2NO→P+N2共释放848.40kJ/mol热量。
在探究金属元素对煤焦燃烧相关反应的影响时,通常采用单一金属原子修饰的方法[23, 25, 26]。本研究中Na的吸附位为焦炭的边缘穴位,吸附模型见图 4(a)。该吸附过程为放热过程,共释放364.04kJ/mol热量。Hirshfeld电荷相较于Mulliken电荷对于电荷迁移的描述更加准确,具体见图 4(b),吸附过程中Na原子向焦炭转移了0.461(a.u.)的电荷,焦炭边缘的原子电荷发生了重排。
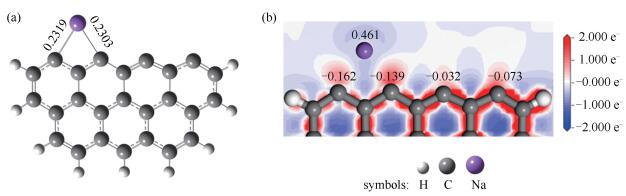
图 4为碱金属Na参与情况下,焦炭异相还原NO反应的反应物、过渡态、中间体及产物的几何构型、主要的键长及过渡态虚频,反应过程的反应势能面则如图 5所示。
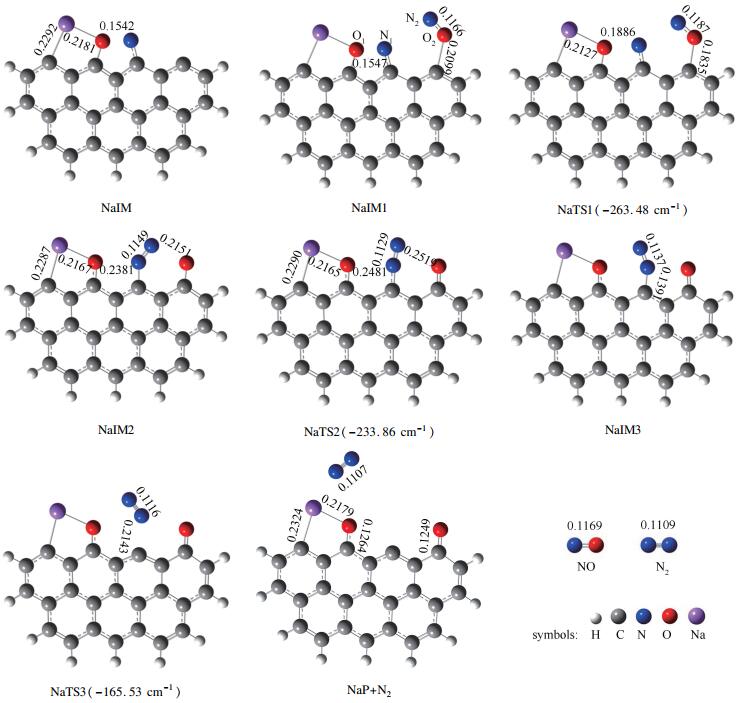
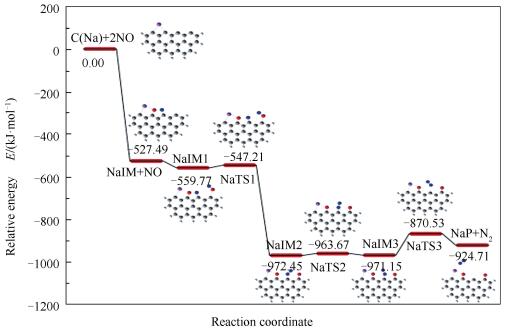
该反应路径需要四个碳活性位。首先一个NO分子同样以side-on模式平行吸附在碳活性位点C2和C3,N1-O1键长由自由状态的0.1169nm伸长至0.1542nm,并释放527.49kJ/mol热量形成NaIM。第二个NO分子以O-down模式吸附于活性位点C4上生成稳定中间体NaIM1,释放出32.27kJ/mol能量。NaIM1翻越12.56kJ/mol能垒变为NaIM2,在此过程中N2-O2键断裂而形成N1-N2键同时有412.68kJ/mol能量放出。N1-N2键绕C3位点逆时针旋转,键长由0.1149nm缩短至0.1137nm,表明键的强度增加;由NaIM2至NaIM3过程翻越8.78kJ/mol能垒。最后,N1-N2键进一步增强,以N2形式由焦炭边缘解吸附,吸收46.44kJ/mol热量并生成产物NaP。最终步即为该反应过程的决速步,其所需活化能为100.62kJ/mol。整个C(Na)+2NO→NaP+N2反应是高放热反应,释放924.71kJ/mol热量。
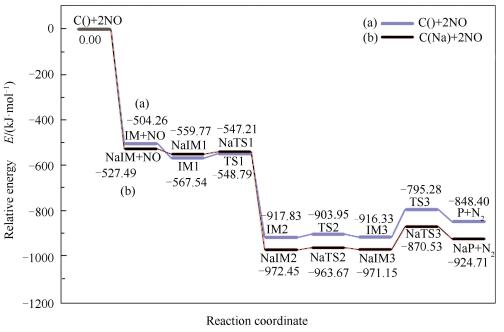
将前述计算的两种反应路径的反应势能面绘制于同一图内,具体见图 7,对比焦炭异相还原NO以及碱金属钠参与的焦炭异相还原NO的反应路径可知,Na的存在强化了焦炭边缘对于第一个NO分子的吸附能力,这与Zhang等[10]的研究一致。Mulliken电荷布局分析可以反映原子与键在吸附前后的电荷分布[27]。
第一个NO分子在两焦炭模型边缘吸附构型的Mulliken电荷布局分析如表 3所示。与NO在焦炭边缘吸附相比,NO吸附于Na存在的焦炭边缘时N1和O1原子的电负性更大,进一步说明了吸附能力更强且结构更加稳定。而焦炭改性后对第二个NO分子系吸附能力没有太大影响。
下载:
导出CSV
| Atom | Charge /(a.u.) | ||
| NO/R | IM | NaIM | |
| N1 | 0.041 | -0.170 | -0.186 |
| O1 | -0.041 | -0.346 | -0.503 |
| C2 | -0.029 | 0.302 | 0.328 |
| C3 | -0.024 | 0.172 | 0.178 |
另一方面,在碱金属Na参与与否的两种情况下,焦炭异相还原NO最大反应能垒均为最终步,即N2在焦炭边缘的脱附过程,该决速步的活化能越小,反应越容易进行。其中,焦炭异相还原NO所需的最大活化能为121.04kJ/mol,碱金属Na参与情况下所需的最大活化能为100.62kJ/mol,即生物质灰中碱金属Na的存在对于焦炭异相还原NO反应有显著的促进作用。
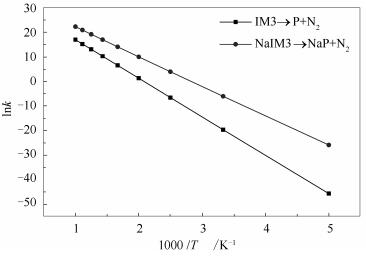
反应动力学计算能够为建立预测模型提供所需的动力学参数。本研究根据经典过渡态理论[19]和决速步理论[28],通过式(1)与式(2)计算了在碱金属钠不参与以及参与情况下焦炭边缘异相还原NO的决速步IM3→P+N2及NaIM3→NaP+N2在温度200-1000 K下的反应速率常数,结果见图 8,相应的动力学参数见表 4。
下载:
导出CSV
| Reaction | Pre-exponential factor A/s-1 | Activation energy Ea/(kJ·mol-1) | Arrhenius equation |
| IM3→P+N2 | 1.50×1014 | 130.29 | k=1.50×1014e-15670.84/T |
| NaIM3→NaP+N2 | 7.32×1014 | 100.20 | k=7.32×1014e-12050.92/T |
由图 8可知,随着温度的上升,两条反应路径的速率常数均增大,该趋势与以往文献结论相吻合[29],说明提高温度对焦炭及碱金属Na参与的焦炭异相还原NO反应均起到促进作用。此外,在所研究温度范围内,碱金属Na参与情况下的焦炭边缘异相还原NO的反应速率常数始终高于单独焦炭边缘的反应速率常数,证明了Na的存在能加快NO在焦炭边缘的还原。由表 4可知,Na参与情况下以及单独焦炭异相还原NO的总反应活化能分别为100.20和130.29kJ/mol,说明NO在Na存在的焦炭边缘的还原反应更易进行。同时,有文献[30]指出指前因子与活性位点的数量有关。由对比可知,碱金属Na参与情况下焦炭异相还原NO反应的指前因子增大,表明焦炭边缘的活性位点数量增多,有利于NO异相还原反应的进行。
本研究采用密度泛函理论并结合经典过渡态理论,针对碱金属钠对焦炭异相还原NO的相关反应的影响机理进行了研究。根据量子化学计算结果可知,碱金属Na参与反应后焦炭对于第一个NO分子的吸附有促进作用,而对于第二个NO分子的吸附影响不大。并且Na的存在不改变反应的过程,其中两条路径的反应决速步均为N2的脱附阶段。动力学分析表明,Na存在的情况下焦炭异相还原NO的总反应活化能更低且反应速率更快,指前因子更大,说明碱金属Na使焦炭边缘的活性位点增多。综上所述,证明了碱金属Na对于焦炭异相还原NO反应具有促进作用。
牛胜利, 韩奎华, 路春美. 生物质先进再燃脱硝特性研究[J]. 燃料化学学报, 2010,38,(6): 745-751. NIU Sheng-li, HAN Kui-hua, LU Chun-mei. Study on nitrogen oxides reduction by advanced biomass reburning process[J]. J Fuel Chem Technol, 2010, 38(6): 745-751.
张国祥, 陈晓晖. 富氧条件下金属催化CO还原NO的研究进展[J]. 化工进展, 2018,37,(12): 4654-4661. ZHANG Guo-xiang, CHEN Xiao-hui. Research progress in metal catalysts for catalytic reduction of NO by CO with excess oxygen[J]. Chem Ind Eng Prog, 2018, 37(12): 4654-4661.
阮丹, 齐砚勇, 李会东. 高温无催化剂条件下CO还原NO数值模拟研究[J]. 硅酸盐通报, 2016,35,(6): 1674-1681. RUAN Dan, QI Yan-yong, LI Hui-dong. Numerical simulation research of the reduction of NO by CO at high temperature without catalyst[J]. Ball Chin Ceram Soc, 2016, 35(6): 1674-1681.
LÓPEZ D, CALO J. The NO carbon reaction:The influence of potassium and CO on reactivity and populations of oxygen surface complexes[J]. Energy Fuels, 2007, 21(4): 1872-1877.
LU P, HAO J T, YU W, ZHU X M, D X. Effects of water vapor and Na/K additives on NO reduction through advanced biomass reburning[J]. Fuel, 2015, 170: 60-66.
WU X, SONG Q, ZHAO H B, YAO Q. Catalytic mechanism of inherent potassium on the Char-NO reaction[J]. Energy Fuels, 2015, 29(11): 7566-7571.
周昊, 刘瑞鹏, 刘子豪, 程明, 岑可法. 碱金属对焦炭燃烧过程中NOx释放的影响[J]. 煤炭学报, 2015,40,(5): 1160-1164. ZHOU Hao, LIU Rui-peng, LIU Zi-hao, CHENG Ming, CEN Ke-fa. Influence of alkali metal on the evolution of NOx during coke combustion[J]. J China Coal Soc, 2015, 40(5): 1160-1164.
刘银河, 刘艳华, 车得福, 徐通模. 煤中灰分和钠添加剂对煤燃烧中氮释放的影响[J]. 中国电机工程学报, 2005,25,(4): 136-141. LIU Yin-he, LIU Yan-hua, CHE De-fu, XU Tong-mo. Effects of minerals and sodium addition on nitrogen release during coal combustion[J]. Proc CSEE, 2005, 25(4): 136-141.
ZHAO Z B, LI W, QIU J S, WANG X Z, LI B Q. Influence of Na and Ca on the emission of NOx during coal combustion[J]. Fuel, 2006, 85(5): 601-606.
ZHANG X X, LIN R Y. Effect of alkali metal elements on nitric oxide chemisorption at the edge of char:A DFT study[J]. Energy Procedia, 2019, 158: 4805-4809.
温正城, 周俊虎, 王智化, 岑可法. 碱金属对煤焦异相还原NO的催化机理:量子化学研究[J]. 浙江大学学报(工学版), 2008,42,(8): 1452-1457. WEN Zheng-cheng, ZHOU Jun-hu, WANG Zhi-hua, CEN Ke-fa. Catalytic mechanism of alkali metal on NO heterogeneous reduction by char:quantum chemistry study[J]. J Zhejiang Univ-Sci(ES), 2008, 42(8): 1452-1457.
PERRY S T, HAMBLY E M, FLETCHER T H, SOLUM M S, PUGMIRE R J. Solid-state 13C NMR characterization of matched tars and chars from rapid coal devolatilization[J]. Proc Combust Inst, 2000, 28(2): 2313-2319.
GAO Z, YANG W, DING X, DING Y, YAN W P. Theoretical research on heterogeneous reduction of N2O by char[J]. Appl Therm Eng, 2017, 126: 28-36.
许紫阳, 岳爽, 王春波, 刘瑞琪. 焦炭催化CO还原NO的反应机理研究[J]. 燃料化学学报, 2020,48,(3): 266-274. XU Zi-yang, YUE Shuang, WANG Chun-bo, LIU Rui-qi. Reaction mechanism of NO reduction with CO catalyzed by char[J]. J Fuel Chem Technol, 2020, 48(3): 266-274.
章勤, 张秀霞, 周俊虎, 周志军, 张彦威, 刘建忠, 岑可法. NO在焦炭表面的吸附特性[J]. 煤炭学报, 2013,38,(9): 1651-1655. ZHANG Qin, ZHANG Xiu-xia, ZHOU Jun-hu, ZHOU Zhi-jun, ZHANG Yan-wei, LIU Jian-zhong, CEN Ke-fa. Characteristics of NO chemisorption on surface of char[J]. J China Coal Soc, 2013, 38(9): 1651-1655.
赵鹏飞, 郭欣, 郑楚光. 活性炭及氯改性活性炭吸附单质汞的机制研究[J]. 中国电机工程学报, 2010,30,(23): 40-44. ZHAO Peng-fei, GUO Xin, ZHENG Chu-guang. Investigating the mechanism of elemental mercury binding on activated carbon and chlorine-embedded activated carbon[J]. Proc CSEE, 2010, 30(23): 40-44.
钟俊, 高正阳, 丁艺, 余岳溪, 杨维结. Zigzag煤焦表面异相还原N2O反应[J]. 煤炭学报, 2017,42,(11): 3028-3034. ZHONG Jun, GAO Zheng-yang, DING Yi, YU Yue-xi, YANG Wei-jie. Heterogeneous reduction reaction of N2O by char based on Zigzag carbonaceous model[J]. J China Coal Soc, 2017, 42(11): 3028-3034.
朱恒毅, 孙保民, 信晶, 尹书剑, 肖海平. 富氧燃烧环境下CO对煤焦异相还原NO的量子化学研究[J]. 煤炭学报, 2015,40,(7): 1641-1647. ZHU Heng-yi, SUN Bao-min, XIN Jing, YIN Shu-jian, XIAO Hai-ping. Quantum chemistry research on NO heterogeneous reduction by char with the participation of CO under oxy-fuel combustion atmosphere[J]. J China Coal Soc, 2015, 40(7): 1641-1647.
ZHANG H, LIU J X, SHEN J, JIANG X M. Thermodynamic and kinetic evaluation of the reaction between NO (nitric oxide) and char(N) (char bound nitrogen) in coal combustion[J]. Energy, 2015, 82: 312-321.
宁华, 刘松, 邓年进, 黎光旭, 蓝志强. Nb(100)表面吸附氮气的第一性原理研究[J]. 广西大学学报(自然科学版), 2013,3,769-776. NING Hua, LIU Song, DENG Nian-jin, LI Guang-xu, LAN Zhi-qiang. Nitrogen adsorption on Nb(100) surface by using First-principles investigation[J]. J Guangxi Univ-Sci(NSE), 2013, 3: 769-776.
李淑萍, 孟江, 王继刚. NO在金属Ben(n=2-12)团簇表面的平行吸附[J]. 原子与分子物理学报, 2019,36,(2): 240-245. LI Shu-ping, MENG Jiang, WANG Ji-gang. Parallel adsorption for NO on the surfaces of Ben (n=2-12) clusters[J]. J At Mol Phys, 2019, 36(2): 240-245.
CHEN N, YANG R T. Ab initio molecular orbital calculation on graphite:Selection of molecular system and model chemistry[J]. Carbon, 1998, 36(7): 1061-1070.
ZHANG X X, XIE M, WU H X, LV X X, ZHOU Z J. DFT study of the effect of Ca on NO heterogeneous reduction by char[J]. Fuel, 2020, 265: .
OYARZúN A M, RADOVIC L R, KYOTANI T. An update on the mechanism of the graphene-NO reaction[J]. Carbon, 2015, 86: 58-68.
GONZÁLEZ J D, MONDRAGÓN F, ESPINAL J F. Effect of calcium on gasification of carbonaceous materials with CO2:A DFT study[J]. Fuel, 2013, 114: 199-205.
ZHAO D, LIU H, SUN C L, XU L F, CAO Q X. DFT study of the catalytic effect of Na on the gasification of carbon-CO2[J]. Combust Flame, 2018, 197: 471-486.
LIU R. Adsorption and dissociation of H2O on Au(111) surface:A DFT study[J]. Comput Theor Chem, 2013, 1019: 141-145.
GAO Z Y, LV S K, YANG W J, YANG P F, MENG X X. Quantum chemistry investigation on the reaction mechanism of the elemental mercury, chlorine, bromine and ozone system[J]. J Mol Model, 2015, 21(6): 2707.
张秀霞.焦炭燃烧过程中氮转化机理与低NOx燃烧技术的开发[D].杭州: 浙江大学, 2012. http://cdmd.cnki.com.cn/Article/CDMD-10335-1012488726.htmZHANG Xiu-xia. Nitrogen conversion mechanism during char combustion and develepment of low NOx technology[D]. Hangzhou: Zhejiang University, 2012. http://cdmd.cnki.com.cn/Article/CDMD-10335-1012488726.htm
易秋明, 刘华财, 阴秀丽, 吴创之. 生物质新生半焦与冷态半焦CO2气化活性差异分析[J]. 新能源进展, 2015,3,9-16. YI Qiu-ming, LIU Hua-cai, YIN Xiu-li, WU Chuang-zhi. Comparative investigation of the CO2 gasification characteristics of biomass In-situ char and Ex-situ char[J]. J Circ Syst, 2015, 3: 9-16.

图 1 Zigzag型焦炭模型示意图
Figure 1 Char model with zigzag edge (carbon atoms at the edge were labeled as: C1, C2, C3, C4)

图 2 NO异相还原反应中间产物及过渡态几何构型(键长:nm)
Figure 2 Geometric parameters for stable species and transition states (bond length: nm)

图 3 Zigzag型焦炭与NO异相还原反应的反应势能面
Figure 3 Reaction potential energy surface of NO heterogeneous reduction at the edge of char

图 4 Na修饰的焦炭模型及其电荷分布
Figure 4 Geometric configuration of Na-decorated char (a) and distribution of Hirshfeld atomic charges (b) (bond length, nm; atomic charge, a. u.)

图 5 Na修饰焦炭异相还原反应中间产物及过渡态几何构型(键长:nm)
Figure 5 Geometric parameters for the stable species and transition states in the NO heterogeneous reduction by Na-decorated char model (bond length: nm)

图 6 Na修饰焦炭与NO异相还原反应过程的反应势能面
Figure 6 Reaction potential energy surfaces of NO heterogeneous reduction at the edge of Na-decorated char
表 1 反应路径各驻点能量及过渡态振动虚频
Table 1. Energies of various compounds and imaginary frequency of transition states
| Species | E/(kJ·mol-1) | Frequency/cm-1 | Species | E/(kJ·mol-1) | Frequency /cm-1 |
| IM | -2856326.30 | - | NaIM2 | -3624341.19 | - |
| IM1 | -3197558.53 | - | NaIM3 | -3624339.89 | - |
| IM2 | -3197908.82 | - | NaP+N2 | -3624293.46 | - |
| IM3 | -3197907.32 | - | NaTS1 | -3623920.36 | -263.48 |
| P+N2 | -3197839.39 | - | NaTS2 | -3624328.91 | -233.86 |
| TS1 | -3197483.39 | -475.11 | NaTS3 | -3624234.95 | -165.53 |
| TS2 | -3197895.36 | -264.72 | NO | -341168.95 | - |
| TS3 | -3197768.49 | -131.19 | N2 | -287649.68 | - |
| NaIM | -3282727.29 | - | R | -2514653.09 | - |
| NaIM1 | -3623928.51 | - | Na | -426013.72 | - |
 下载: 导出CSV
下载: 导出CSV
表 2 模型平均键长的计算值和文献值对比
Table 2. Calculated average bond length for the model used in this work, compared with those reported in the literature
下载: 导出CSV
表 3 NO在焦炭及Na修饰焦炭边缘吸附的Mulliken电荷布局分析
Table 3. Mulliken atomic charge populations for NO adsorption on the edge of char and Na-decorated char
| Atom | Charge /(a.u.) | ||
| NO/R | IM | NaIM | |
| N1 | 0.041 | -0.170 | -0.186 |
| O1 | -0.041 | -0.346 | -0.503 |
| C2 | -0.029 | 0.302 | 0.328 |
| C3 | -0.024 | 0.172 | 0.178 |
下载: 导出CSV
表 4 反应动力学参数
Table 4. Reaction kinetic parameters
| Reaction | Pre-exponential factor A/s-1 | Activation energy Ea/(kJ·mol-1) | Arrhenius equation |
| IM3→P+N2 | 1.50×1014 | 130.29 | k=1.50×1014e-15670.84/T |
| NaIM3→NaP+N2 | 7.32×1014 | 100.20 | k=7.32×1014e-12050.92/T |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们