图式 1.
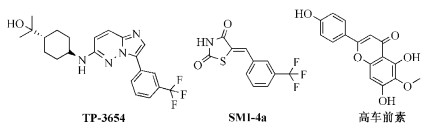
化合物TP-3654、SMI-4a、高车前素的结构式
Scheme 1.
Structure of TP-3654, SMI-4a, hispidulin as Pim-1 kinase inhibitors

癌症是21世纪世界各国人口死亡的主要原因之一,极大地危害人类的生存和健康。2018年,全世界新增1810万例癌症患者,并且有960万例癌症患者死亡[1]。癌症的出现及发展与信号转导通路密切相关,针对细胞信号转导通路中的各种关键因子开发特异性分子靶标的高效低毒药物成为抗肿瘤药物研发的重点[2]。蛋白激酶在细胞信号通路中发挥着重要作用,它能将蛋白质磷酸化,进而起调控蛋白质功能的作用[3]。目前许多抗癌药物研究聚焦在小分子蛋白激酶抑制剂开发上,同时有很多临床在用小分子蛋白激酶抑制剂,如伊马替尼和吉非替尼等[4]。
Pim激酶是一种丝氨酸/苏氨酸蛋白激酶,已发现Pim-1、Pim-2和Pim-3三个成员,其60%~70%的氨基酸序列一致,均由原癌基因Pim编码[5]。Pim激酶已被发现在恶性血液病及许多实体瘤的发生发展中过度表达,可通过多种不同机制影响肿瘤细胞的增殖,如促进细胞周期G1/S期的进程,正向调节STAT3、Wnt信号传导通路,也可正向调节NF-κB信号传导系统,从而促进细胞增殖[6]。
Pim-1激酶在许多实体肿瘤中过表达,其中包括胰腺癌、前列腺癌、胃癌、结直肠癌、肝癌以及膀胱癌等,而Pim-2、Pim-3激酶只在少数肿瘤细胞中过表达[7]。Pim-1激酶通过作用于下游靶点参与细胞周期进程和细胞凋亡的调节,影响肿瘤的发生发展。研究表明,Pim-1激酶通过作用于多种信号通路如GSK3B、FOXP3促进前列腺癌细胞转移。另外,Pim-1激酶能够磷酸化P-糖蛋白、乳腺癌耐药蛋白,从而使它们在细胞表面起到药物流出泵的作用[8~10]。研究表明,缺失Pim-1激酶的小鼠也可以正常生长,且不产生关键性副作用[11]。因此,Pim-1激酶在肿瘤研究领域中被认为是良好的潜在靶标,多种结构类型的小分子Pim-1激酶抑制剂处于不同研究阶段中[8]。例如,TP-3654(图式 1)以吡唑[1, 5-a]嘧啶为活性骨架,在Pim-1/BAD过表达系统中显示出有效的Pim-1特异性细胞活性,在小鼠模型实验中,TP-3654能够抑制小鼠皮下肿瘤的生长,并且没有显著毒性[12],目前该抑制剂处于Ⅰ期临床研究阶段。SMI-4a是苯亚甲基-噻唑烷-2, 4-二酮类小分子化合物,可在体内和体外选择性抑制Pim-1激酶的活性,其IC50值为24nmol/L[13]。天然黄酮类化合物高车前素能很好地固定于Pim-1激酶的ATP结合缝隙中,其作为Pim-1激酶抑制剂目前处于临床前研究阶段[14]。
本文采用Topomer CoMFA方法建立3, 5-二取代的6-氮杂吲唑类Pim-1激酶抑制剂的3D-QSAR模型,并建立该类化合物的药效团模型,总结活性化合物共同药效特征信息。基于所建上述模型,进行新分子设计及分子对接研究,从而验证本文所建模型的可靠性,为新型Pim-1激酶抑制剂的设计合成提供设计指导和理论依据。
采用Tripos公司SYBYL-X2.1.1软件包进行研究:采用“Minimize”功能模块对分子结构进行优化,得到合理的构象;采用“Topomer CoMFA”功能模块建立3D-QSAR模型,并使用所建模型对化合物的活性进行预测;采用“GALAHAD”功能模块建立药效团模型,探究与生物活性关系密切的药效团特征元素及其空间分布;采用“Surflex-Dock”功能模块完成受体大分子与配体小分子的对接,探索两者间的作用模式。
本文选取的39个3, 5-二取代的6-氮杂吲唑类Pim-1激酶抑制剂来自于文献报道[15],在3D-QSAR模型建立过程中,将39个化合物依据活性梯度排序后,按随机原则抽取31个化合物作为训练集建立3D-QSAR模型,另外8个化合物作为测试集用于测试已建立模型的预测能力。39个化合物的结构及活性测试结果见表 1。本文中使用抑制常数Ki值(反映抑制剂对靶标的抑制强度)的常用对数的负值(即-lgKi,简称pKi)作为活性数据。
 下载:
导出CSV
下载:
导出CSV

|
采用“Minimize”功能模块完成39个化合物结构的分子力学能量优化。优化过程选用Powell能量梯度法,使用Tripos力场,加载Gasteiger-Hückel电荷,对所有化合物分子进行优化,最大重复次数为1000次,能量收敛条件为能级差0.001kcal/(mol· Å),其他参数均采用软件默认值。
采用“Topomer CoMFA”功能模块对训练集中31个化合物进行切割,以化合物17作为模板分子,对训练集分子进行切割,切割方式如图 1所示,分别生成R1(蓝色)和R2(红色)基团。随后,软件自动识别并且切割其他分子结构,最后对R1和R2基团周围的立体场和静电场自动进行计算,并把它作为自变量,以Ki的负对数(pKi)为建模响应值。采用偏最小二乘(PLS)回归分析法进行建模来表示化合物活性与分子场之间的关系,生成3D-QSAR模型,结合pKi活性数据与Topomer CoMFA所得立体场(Steric,S)、静电场(Electrostaic,E)进行模型分析。采用留一法(Leave One-Out,LOO)交互验证评价模型的内部预测能力[16],经过计算可得最佳主成分数N和交叉验证系数q2,通过非交叉验证法建立3D-QSAR模型预测活性数据,并得到非交叉验证系数(r2)、标准偏差(SEE)和显著系数(F)。利用所建立的3D-QSAR模型预测8个测试集化合物的活性,以此评价模型的外部预测能力。另外,为了进一步评价模型的可靠性,还必须运用不同统计量及方法对所建模型进行外部验证,其中包括Golbraikh-Tropsha统计方法和统计量QF12、QF22、QF32。外部验证相关统计量的计算公式见表 2。
 下载:
导出CSV
下载:
导出CSV
| 统计量名称 | 数学等式 |
| Golbraikh和Tropsha方法 | $R=\frac{\sum_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}_{E X T}\right)\left(\hat{y}_{i}-\overline{\hat{y}}\right)}{{\sqrt{\sum_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}_{E X T}\right)^{2} \sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-\hat{y}\right)^{2}}}}$ $k=\sum_{i=1}^{n_{E X T}} y_{i} \hat{y}_{i} / \sum_{i=1}^{n_{E X T}} \hat{y}_{i}^{2}, y_{i}^{r o}=k \hat{y}_{i}$ $k^{\prime}=\sum_{i=1}^{n_{E X T}} y_{i} \hat{y}_{i} / \sum_{i=1}^{n_{E X T}} y_{i}^{2}, \hat{y}_{i}^{r o}=k^{\prime} y_{i}$ $R_{o}^{2}=1-\sum\limits_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}^{r o}\right)^{2} / \sum\limits_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-\overline{{\hat{y}}}\right)^{2}$ $R_{o}^{\prime 2}=1-\sum\limits_{i=1}^{n_{E X T}}\left(y_{i}-\hat{y}_{i}^{r o}\right)^{2} / \sum\limits_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}\right)^{2}$ |
| QF22 | $Q_{F 1}^{2}=1-\frac{\sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}\right)^{2}}{\sum_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}_{T R}\right)^{2}}$ |
| QF22 | $Q_{F 2}^{2}=1-\frac{\sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}\right)^{2}}{\sum_{i=1}^{n_{E X T}\left(y_{i}-\bar{y}_{E X T}\right)^{2}}}$ |
| QF32 | $Q_{F 3}^{2}=1-\frac{\sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}\right)^{2} / n_{E X T}}{\sum_{i=1}^{n_{TR}}\left(y_{i}-\bar{y}_{T R}\right)^{2} / n_{T R}}$ |
| 注:n是样本数;nEXT和nTR分别是测试集和训练集的样本数;yi是实验值;y是实验值的平均值;yEXT是测试集实验值的平均值;yTR是训练集实验值的平均值;ŷi是预测值;ŷ是预测值的平均值;k和k′是斜率;Ro2是预测对实验活性的相关系数;R′o2是实验对预测活性的相关活性。 | |
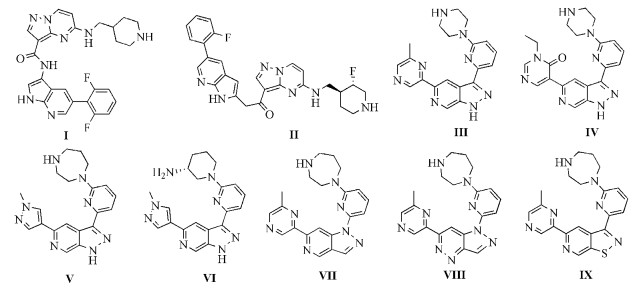
用于建立药效团模型的训练集分子Ⅰ、Ⅱ来源于文献[17],Ⅲ~Ⅵ来源于文献[15],Ⅶ~Ⅸ来源于文献[18],具体结构见图式 2。在GALAHAD操作界面中,将“Population size”值设置为55;“Max generations”值设置为40;“Mols required to hit”值设置为4,其他参数均为默认值。选择Specificity,N_hits,Features和Mol_Qry值最大的模型为最优药效团模型。将最优药效团模型提取UNITY提问式,设置UNITY搜寻选项,“Query Type”设置为“Flex Search”;“Use Lipinski’s ‘rule of 5’”设为“No”。将3D-QSAR建模所选39个分子为测试集导入,检验最优药效团模型的有效性。
采用“Surflex-Dock”功能模块进行15个新设计的3, 5-二取代的6-氮杂吲唑类化合物(表 3)与Pim-1、Pim-2激酶的分子对接。对接时所用的Pim-1、Pim-2激酶三维晶体复合物结构来源于数据库Protein Data Bank(PDB,http://www.rcsb.org),编号分别为5DIA、4X7Q。以化合物与Pim-1激酶分子对接为例,在蛋白质修饰过程中,抽离出原配体5E6(即表 1中的化合物35),去除所有水分子,将已抽离配体的蛋白质进行加氢、指定AMBER7-99的原子类型、加载Gasteiger-Hückel电荷、修正错误或不完整残基等操作。以5E6的分子结构为参照,进行新设计的3, 5-二取代的6-氮杂吲唑类化合物与Pim-1激酶的对接,结合部位为5DIA中Pim-1激酶与5E6的结合部位。Total-score为给出的总打分函数,评分越高者与受体亲和力越强,以评分值较高者作为较优分子构象;Crash代表配体对接近受体时的不适当程度;Polar为极性打分函数;Similarity代表分子构象相似性;C-score为配体—受体亲和力的一致性评分函数。最后,使用PyMOL软件[19]生成对接结果的图形视图。
下载:
导出CSV

|
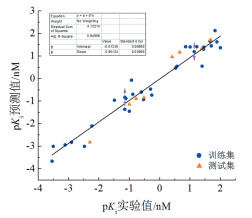
通常认为,当q2>0.5,r2>0.8时3D-QSAR模型具有较好的内部预测能力,利用该模型预测得到的化合物活性结果可靠[20]。本文所建模型的线性方程为y=0.9512x-0.0134(x为实验实际测得的活性数值;y为模型预测活性数值),q2=0.756,r2=0.951,N=5,SEE=0.37,F=273.11。结果表明,所建3D-QSAR模型具有可靠的预测活性的能力,能够较为准确的预测测试集化合物活性。对于本文引入的外部预测能力评价方法,计算所得R=0.83、k=0.82、k′=1.17、R02=0.97、R′02=0.97,满足Golbraikh-Tropsha方法[21]提出的条件:预测与实际活性之间的相关系数R应接近于1;通过原点的回归线斜率应接近于1;相关系数R02和R′02应满足(R2-R02)/R2 < 0.1或(R2-R′02)/R2 < 0.1。另外,统计量QF12=0.92、QF22=0.91、QF32=0.96,符合Chirico等[22]建议外部统计量的阈值为QF12、QF22和QF32>0.6。以上数据证明该3D-QSAR模型具有较强的外部预测能力。3D-QSAR模型中Pim-1激酶抑制剂的pKi实验值与预测值相关性曲线见图 2,表示化合物的点均分布在直线两侧,进一步表明模型具有良好稳定性及预测能力,该模型用于预测新化合物活性具有较高可信度。
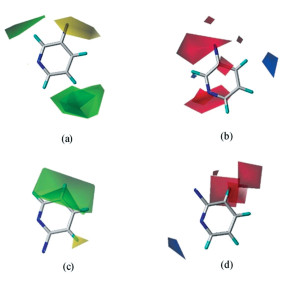
以训练集中中等活性的化合物8为例,进一步分析取代基团对于提高或降低化合物活性的具体影响。图 3为化合物8的Topomer CoMFA模型的分子场三维等势图。图 3(a)、(c)分别是R1,R2基团立体场计算结果,在绿色区域引入大体积基团,在黄色区域引入小体积基团有助于提高化合物活性,反之活性降低。图 3(b)、(d)显示的是Topomer CoMFA模型的静电场计算结果,在蓝色区域引入带正电荷基团,红色区域引入带负电荷基团有助于提高化合物活性。图 3(b)中显示有大片的红色等势区域,说明此处引入负电荷基团有助于提高化合物活性,与化合物8相比,化合物13在此处引入4-氨基哌啶基,使化合物活性提高;化合物15是在化合物8的R2基团上引入哌嗪基,恰好位于图 3(c)中的绿色区域等势图,与此同时,该哌嗪基也与图 3(d)中的红色区域等势图相符,因此化合物15活性比化合物8的活性显著提高。
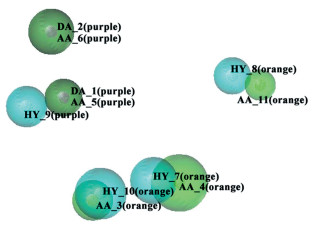
GALAHAD药效团模块研究所得最优药效团模型见图 4,黄色标签表示所选取的分子具有同一个药效团,橙色标签表示选取的大部分分子具有同一个药效团,蓝绿色球体代表疏水团、绿色球体代表氢键受体、品红色球体代表氢键供体。从图 4可以看到,药效团模型中有4个疏水团(HY_7、HY_8、HY_9、HY_10),5个氢键受体(AA_3、AA_4、AA_5、AA_6、AA_11)与2个氢键供体(DA_1、DA_2)。AA_3、AA_4、AA_11、HY_7、HY_8、HY_10等这6个药效团是大部分分子都存在。四个疏水团存在的位置是Pim-1激酶抑制剂的骨架上6-氮杂吲唑环、嘧啶环或吡啶环;代表氢键受体的2个药效团AA_3、AA_4和代表疏水团的2个药效团HY_7、HY_10位于6-氮杂吲唑环上,说明母核结构为具有氢键受体的芳香杂环对化合物活性有利;氢键受体AA_11、疏水团HY_8两个药效团位置接近,观察所选的9个化合物,发现在此位置多数化合物带有含氢键受体的芳香杂环结构,这表明此位置引入芳香杂环取代基,对化合物的活性有所帮助。最后,利用此药效团模型对图式 2中39个分子进行筛选,除化合物4、7外,命中37个活性分子,表明该药效团模型能够准确反映6-氮吲唑类Pim-1激酶抑制剂的结构特征,可用于今后Pim-1激酶抑制剂的筛选及设计。
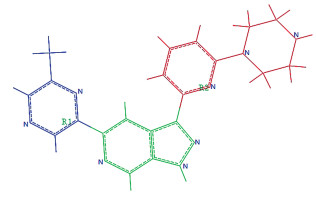
基于上述研究获得的3D-QSAR模型以及药效团模型提供的指导信息,通过变换模板分子17的R1和R2基团,共设计了15个新型Pim-1激酶抑制剂,并利用上文Topomer CoMFA所建模型对15个新分子进行活性预测,同时进行了分子对接研究。新分子结构及其活性预测值与分子对接打分函数值见表 3。由表中数据可知,新设计分子的Pim-1激酶抑制预测活性高于大多数建模分子,表明3D-QSAR所建模型可为新型抑制剂的设计提供可靠的理论指导。尤其是化合物b、i、n、o的预测pIC50值高于原文献活性最好的化合物17,有望成为新型Pim-1激酶抑制剂候选化合物,值得进一步研究。从结构中可知,对Pim-1激酶的抑制活性最强的化合物17,b、i、n、o的R1取代基分别为2-甲基吡嗪基、N1-(3-吡啶基)乙二胺基、3-吡啶基乙腈基,R2取代基分别为1-(2-吡啶基)哌嗪基或1-[5-(三氟甲基)-2-吡啶基]哌嗪基。按照3D-QSAR模型立体场及静电场要求且保留药效团模型中对活性贡献大的元素,与化合物17相比,在化合物b和i的R1取代基相应位置分别引入了大体积且电正性基团乙二胺基或乙腈基,使其活性得到提高。另外化合物l~o均在R2取代基相应位置引入了电负性强的三氟甲基,从而使其活性高于多数训练集化合物。因此,3D-QSAR模型立体场及静电场等势结果都可成为将来新型Pim-1激酶抑制剂设计及改造的重点考虑方向。
在对以上新设计分子进行分子对接之前,将蛋白晶体结构5DIA中的原配体5E6(即表 1中的化合物35)抽离出来,然后重新将化合物35与其进行对接,以对本文分子对接方法及其准确性进行验证。另外,选取原文献中活性最高的化合物17与蛋白进行对接,将其对接结果作为对照。图 5(a)是化合物35与Pim-1激酶上氨基酸的具体作用模式,其共与2个氨基酸发生氢键相互作用,形成2个氢键。其中6-氮杂吲唑环1位的NH作为氢键供体,与氨基酸残基GLU121的羰基氧形成1个氢键,氢键距离为2.0 Å;吡啶环的N作为氢键受体与LYS67的氨基形成1个氢键,氢键距离为2.4 Å,该结果与原文献[15]一致,表明本对接方法可以实现结合空腔位置的准确定位,可以进行有效的分子对接研究。化合物17的对接结果见图 5(b),其6-氮杂吲唑环1位的NH与化合物35以相同的方式与相同的氨基酸残基发生氢键作用;嘧啶环上的N原子与LYS67的氨基之间形成了氢键,Total-score为7.318。
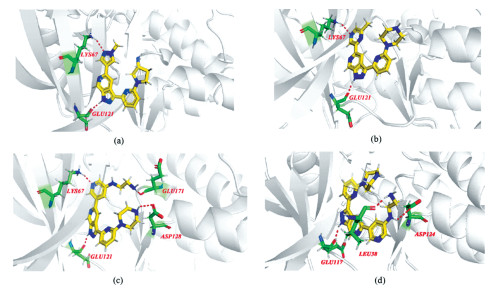
15个新设计的Pim-1激酶抑制剂与Pim-1激酶对接打分详见表 3。在对接结果中,所有化合物均很好地伸入蛋白的活性口袋中,有利于其更好地发挥对蛋白的抑制作用,且所有化合物均与关键氨基酸残基GLU121和LYS67发生氢键相互作用。其中,化合物b得分最高(Total-score=10.240)。图 5(c)是化合物b与Pim-1激酶上氨基酸的具体作用模式。从图中可以看出,化合物b共与4个氨基酸残基发生氢键相互作用,形成4个氢键。其中,与氨基酸残基GLU121、LYS67的相互作用方式与化合物17一致,氢键距离分别为2.1和2.0 Å。而其R1基团引入的大体积基团中末端氨基上的H原子与氨基酸残基GLU171的羰基O原子形成1个新的氢键(1.9 Å)。另外,可能由于R1基团空间位阻使其R2基团较化合物17发生了构象改变,使R2基团上的哌嗪环NH与氨基酸残基ASP128形成一个新的氢键(2.4 Å)。因此新设计的化合物b与Pim-1激酶的适配程度、结合亲和力、稳定性及活性高于化合物17,该结论与3D-QSAR立体场等势结果一致,进一步说明了本对接方法的有效性和可靠性。结合15个新设计分子所得的较高预测活性及其与靶蛋白Pim-1激酶优良的对接结果,表明本对接方法也可为将来新型Pim-1激酶抑制剂设计及改造提供参考。此外,Ishchenko等[23]的研究表明,Pim-1与Pim-2活性位点处的氨基酸具有90%的序列相似性,导致化合物对两种激酶活性差别不大。将15个新设计分子与Pim-2激酶进行对接,所有分子均较好地结合在Pim-2激酶的活性口袋中。其中,化合物b对接打分最高,其与Pim-2激酶上氨基酸的具体作用模式见图 5(d)。从图中可知,化合物b同样可以深入到Pim-2激酶的活性口袋中,且其6-氮杂吲唑环1位的NH与氨基酸残基GLU117的羰基氧形成的氢键相互作用与Pim-1激酶相同。Pim-3激酶的晶体结构尚未见报道,但根据序列相似性推测所设计的Pim-1激酶抑制剂可能可以同时抑制Pim-3激酶。因此,基于本文所建模型设计的新分子可能同时具有抑制三种Pim激酶的作用。
本文联合使用3D-QSAR与构建药效团模型两种计算方法,对39个Pim-1激酶抑制剂进行计算机辅助药物设计模拟计算。将39个化合物按照活性梯度排序后随机抽选了31个化合物,经过计算建立了q2=0.756,r2=0.951的Topomer-CoMFA模型。模型经过内外部预测,计算各种模型检验参数,以确定该3D-QSAR模型具有较好预测能力及稳定性,并可通过立体场、静电场三维等势图以直观形式进行分析,在R1基团中引入大体积、正电性基团,在R2基团中引入大体积、负电性基团能明显增强抑制作用,该项3D-QSAR研究可用于指导6-氮杂吲唑类Pim-1激酶抑制剂的结构优化。通过药效团模型可以发现,母核为含有氢键受体的芳香杂环结构,另外侧链取代基含有芳香杂环结构的化合物能够具有很好的Pim-1激酶抑制作用。此外,基于等势图和药效团模型所提供的信息,新设计了15个Pim-1激酶抑制剂分子,运用所建3D-QSAR模型预测均具有较高活性,进一步确认本文所建3D-QSAR模型的可靠性。且分子对接模式研究显示新设计分子均与靶蛋白Pim-1、Pim-2激酶具有较高结合亲和力,对接结果与3D-QSAR等势图结果一致,表明该分子对接方法有效可靠,并可将该对接方法用于新型Pim激酶抑制剂的设计。因此,本研究中3D-QSAR模型与药效团模型的建立,可以对未来新型Pim-1激酶抑制剂的设计提供一定的帮助和支持。
马丹丹, 刘坤, 齐晓伟.中华乳腺病杂志(电子版), 2018, 12(06): 375~375.
方罗, 吴盈盈, 张翀, 等.中国肿瘤, 2013, 22(8): 644~655.
Tarrant M K, Cole P A. Annu. Rev. Biochem., 2009, 78(01): 797~825.
郑培根, 郑程隆.农产品加工, 2016, 1: 49~52.
刘浩秋.中国当代医药, 2014, 21(28): 191~193.
郑小洲, 金晓磊, 赵临襄.中国药物化学杂志, 2013, 23(6): 499~505.
Narlik-grassow M, Blanco-aparicio C, Carnero A. Med. Res. Rev., 2014, 34(1): 136~159. doi: 10.1002/med.21284
周育奇, 龚晓峰, 全兴萍, 等.中国新药杂志, 2019, 28(1): 49~53.
Santio N M, Salmela M, Arola H, et al. Exp. Cell. Res., 2016, 342(02): 113~124. doi: 10.1016/j.yexcr.2016.02.018
Xie Y, Burcu M, Linn D E, et al. Mol. Pharmacol., 2010, 78(02): 310~318. doi: 10.1124/mol.109.061713
Larid P W, Vanderlugt N M T, Clarke A, et al. Nucl. Acids Res., 1993, 21(20): 4750~4755. doi: 10.1093/nar/21.20.4750
Foulks J M, Carpenter K J, Luo B, et al. Neoplasia, 2014, 16(05): 403~412. doi: 10.1016/j.neo.2014.05.004
Lin Y W, Beharry Z M, Hill E G, et al. Blood, 2010, 115(04): 824~833. doi: 10.1182/blood-2009-07-233445
Chao S W, Su M Y, Chiou L C, et al. J. Nat. Prod., 2015, 78(08): 1969~1976. doi: 10.1021/acs.jnatprod.5b00324
Hu H, Wang X, Chan G K Y, et al. Bioorg. Med. Chem. Lett., 2015, 25(22): 5258~5264. doi: 10.1016/j.bmcl.2015.09.052
Yu S, Yuan J, Shi J, et al. Chemom. Intell. Lab. Syst., 2015, 146: 34~41. doi: 10.1016/j.chemolab.2015.04.017
Wang X, Magnuson S, Pastor R, et al. Bioorg. Med. Chem. Lett., 2013, 23(11): 3149~3153. doi: 10.1016/j.bmcl.2013.04.020
Wang X, Kolesnikov A, Tay S, et al. J. Med. Chem., 2017, 60(10): 4458~4473. doi: 10.1021/acs.jmedchem.7b00418
The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
Niu B, Lu Y, Wang J Y, et al. Comput. Struct. Biotechnol. J., 2019, 17: 36~48.
Golbraikh A, Tropsha A. J. Mo. Graphics. Modell., 2002, 20(4): 269~276. doi: 10.1016/S1093-3263(01)00123-1
Chirico N, Gramatica P. J. Chem. Inf. Model., 2011, 51(9): 2320~2335. doi: 10.1021/ci200211n
Ishchenko A, Zhang L, LeBrazidec J Y, et al. Bioorg. Med. Chem. Lett., 2015, 25(3): 474~480. doi: 10.1016/j.bmcl.2014.12.041

图式 1 化合物TP-3654、SMI-4a、高车前素的结构式
Scheme 1 Structure of TP-3654, SMI-4a, hispidulin as Pim-1 kinase inhibitors

图 1 Topomer CoMFA建模中化合物17的切割方式
Figure 1 Cutting method of compound 17 in Topomer CoMFA modeling

图 2 Topomer CoMFA模型实测值与预测值的相关性
Figure 2 Correlation of experimental value and predicted value of Topomer CoMFA model

图 3 化合物8的Topomer CoMFA模型立体场(a,c)与静电场(b,d)等势图
Figure 3 Stereo field (a), (c) and electrostatic field (b), (d) isopotential diagram of compound 8 in Topomer CoMFA model

图 5 化合物35(a)、17(b)、b(c)与Pim-1激酶及化合物b(d)与Pim-2激酶氨基酸残基结合模式图
Figure 5 Binding pattern of compounds 35(a), 17(b) and b(c) with amino acid residues of Pim-1 kinase and compound b(d) with amino acid residues of Pim-2 kinase
表 1 39个Pim-1激酶抑制剂化合物结构及活性测试结果
Table 1. Structures and biological activity results of 39 Pim-1 kinase inhibitor compounds
|
|
 下载: 导出CSV
下载: 导出CSV
表 2 3D-QSAR模型外部检验统计量计算公式
Table 2. Statistics calculation formulas of external test of 3D-QSAR Model
| 统计量名称 | 数学等式 |
| Golbraikh和Tropsha方法 | $R=\frac{\sum_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}_{E X T}\right)\left(\hat{y}_{i}-\overline{\hat{y}}\right)}{{\sqrt{\sum_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}_{E X T}\right)^{2} \sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-\hat{y}\right)^{2}}}}$ $k=\sum_{i=1}^{n_{E X T}} y_{i} \hat{y}_{i} / \sum_{i=1}^{n_{E X T}} \hat{y}_{i}^{2}, y_{i}^{r o}=k \hat{y}_{i}$ $k^{\prime}=\sum_{i=1}^{n_{E X T}} y_{i} \hat{y}_{i} / \sum_{i=1}^{n_{E X T}} y_{i}^{2}, \hat{y}_{i}^{r o}=k^{\prime} y_{i}$ $R_{o}^{2}=1-\sum\limits_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}^{r o}\right)^{2} / \sum\limits_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-\overline{{\hat{y}}}\right)^{2}$ $R_{o}^{\prime 2}=1-\sum\limits_{i=1}^{n_{E X T}}\left(y_{i}-\hat{y}_{i}^{r o}\right)^{2} / \sum\limits_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}\right)^{2}$ |
| QF22 | $Q_{F 1}^{2}=1-\frac{\sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}\right)^{2}}{\sum_{i=1}^{n_{E X T}}\left(y_{i}-\bar{y}_{T R}\right)^{2}}$ |
| QF22 | $Q_{F 2}^{2}=1-\frac{\sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}\right)^{2}}{\sum_{i=1}^{n_{E X T}\left(y_{i}-\bar{y}_{E X T}\right)^{2}}}$ |
| QF32 | $Q_{F 3}^{2}=1-\frac{\sum_{i=1}^{n_{E X T}}\left(\hat{y}_{i}-y_{i}\right)^{2} / n_{E X T}}{\sum_{i=1}^{n_{TR}}\left(y_{i}-\bar{y}_{T R}\right)^{2} / n_{T R}}$ |
| 注:n是样本数;nEXT和nTR分别是测试集和训练集的样本数;yi是实验值;y是实验值的平均值;yEXT是测试集实验值的平均值;yTR是训练集实验值的平均值;ŷi是预测值;ŷ是预测值的平均值;k和k′是斜率;Ro2是预测对实验活性的相关系数;R′o2是实验对预测活性的相关活性。 | |
下载: 导出CSV
表 3 新设计Pim-1激酶抑制剂分子的结构、活性预测值及对接打分值
Table 3. Structures, predictive activity values and docking scores of new designed Pim-1 kinase inhibitor molecules
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们