 图 1
以Ce(NO3)3-Cu3(BTC)2为前驱体制备的CuO/CeO2催化CO氧化[46]
Figure 1.
CuO/CeO2 prepared from Ce(NO3)3-Cu3(BTC)2 precursor for preferential CO oxidation[46]
图 1
以Ce(NO3)3-Cu3(BTC)2为前驱体制备的CuO/CeO2催化CO氧化[46]
Figure 1.
CuO/CeO2 prepared from Ce(NO3)3-Cu3(BTC)2 precursor for preferential CO oxidation[46]
引用本文:
王丽苹, 赵粒成, 兰开顺. 以MOFs为前驱体的多孔金属氧化物催化剂研究进展[J]. 化学通报,
2017, 80(7): 611-620.

Citation: Wang Liping, Zhao Lichen, Lan Kaishun. Progress in Porous Metal Oxide Catalysts Derived from MOFs[J]. Chemistry, 2017, 80(7): 611-620.
Citation: Wang Liping, Zhao Lichen, Lan Kaishun. Progress in Porous Metal Oxide Catalysts Derived from MOFs[J]. Chemistry, 2017, 80(7): 611-620.
以MOFs为前驱体的多孔金属氧化物催化剂研究进展
摘要:
多孔金属氧化物具有高比表面积、大孔径、特殊的形貌和结构特性,广泛应用于催化、锂离子电池、太阳能电池、气敏传感器等领域。金属有机骨架材料(MOFs)是一类具有周期性网络结构的新型多孔晶体材料,在气体存储、气体分离、催化等领域具有重要的应用价值。近年来,以MOFs为前驱体制备多孔碳和多孔金属氧化物成为MOFs应用领域一个新的研究热点。本文主要综述了以MOFs为前驱体制备的多孔金属氧化物和多孔金属氧化物/碳复合物在CO氧化、催化产氢、异丁烷脱氢、环已烯氧化、醇直接氧化为酯、醛氧化酰胺化反应、光催化降解有机物和氧还原反应等方面的应用。
English
Progress in Porous Metal Oxide Catalysts Derived from MOFs
Abstract:
Porous metal oxides are used widely in catalysis, lithium ion battery, solar cell and gas sensor due to their high surface areas, large pore size, special morphology and structure characteristics. As a novel class of porous crystalline materials with periodic network structure, metal-organic frameworks (MOFs) are widely applied in gas storage, gas separation and catalysis. In recent years, the preparation of porous carbon and porous metal oxide using MOFs as precursors has become a new research hotspot in MOFs applications. This work reviews that porous metal oxides and metal oxides/carbon composites derived from MOFs are used as the catalysts for CO oxidation, hydrogen production, dehydrogenation of isobutane, oxidation of cyclohexene, direct oxidation of alcohols to esters, oxidative amidation of aldehydes, degradation of organic compounds and oxygen reduction.
-
Key words:
- Metal-organic framework
- / Porous metal oxide
- / Calcination
- / Catalyst
- / Catalytic activity
-
凭借其独特的物理和化学性质,金属氧化物纳米材料在催化[1, 2]、传感器[3, 4]、能源[5, 6]等领域展现出极大的应用价值。金属氧化物催化剂在烃类的选择性氧化[7]、NOx还原[8]、加氢反应[9]、光催化[10]、CO氧化[11]等反应中展现出优异的催化活性。多孔金属氧化物具有发达的孔道结构、高的比表面积、特殊的形貌和结构特性,有利于反应物分子的扩散、吸附和活化,因而具有更高的催化活性[12]。Li等[13, 14]报道了介孔Co3O4和Au/介孔Co3O4在乙烯氧化反应中的催化性能。与Co3O4纳米薄膜相比,具有高活性晶面和孔结构的介孔Co3O4在乙烯氧化反应中显示出更加优良的催化活性能。介孔Co3O4的孔道结构有利于反应物和产物的传输和Au纳米粒子的分散,有效防止了Au纳米粒子的团聚和流失。陈英[15]考察了负载不同活性组分(V2O5、WO3、CuO、Al2O3)的介孔ZrO2-TiO2复合氧化物在NOx还原反应中的催化活性。结果表明,介孔复合氧化物催化剂在NOx还原反应中的催化活性明显优于商业催化剂V2O5-WO3/TiO2。Chen等[16]研究了介孔Nb2O5在光催化水分解制氢反应中的催化性能。结果显示,介孔Nb2O5的光催化活性是纳米Nb2O5活性的20倍。Luo等[17]将Pd/介孔Co3O4-CeO2复合氧化物用作低温氧化CO的催化剂,获得了良好的实验结果,复合物氧化物的多孔结构和高比表面积有利于Pd纳米粒子的分散。由此可见,多孔金属氧化物在催化领域的应用受到了人们越来越多的关注[18]。
前驱体热分解法制备多孔金属氧化物具有操作简便的特点,且焙烧产物与前驱体具有相同的形貌,结构不易被破坏。Liu等[19]将水热法获得的Zn5(CO3)2(OH)6在空气中下焙烧得到多孔ZnO。ZnO保留了前驱体的形貌,BET比表面积为12.5m2/g,孔体积为0.041cm3/g。Sinhamahapatra等[20]利用沉淀法制备前驱体Zn5(CO3)2(OH)6,所得ZnO的BET比表面积和孔体积得到很大提高,分别为44m2/g和0.19cm3/g。前驱体热分解法制备多孔金属氧化物的关键就是前驱体的制备。金属有机骨架材料(MOFs)是一类由有机配体与金属或金属簇通过自组装过程形成的具有周期性网络结构的新材料,具有可调控的孔尺寸、可修饰的孔道表面、高比表面积等独特的性能优势[21]。因此,MOFs在气体存储[22, 23]和催化[24, 25]等方面得到广泛应用。MOFs凭借其丰富的活性位点,常用作烷基化反应[26]、氧还原反应[27]、催化加氢[28]、偶联反应[29]、酯交换反应[30~33]、氧化反应[34~36]等的催化剂。近年来,随着MOFs材料设计和制备技术的快速发展,大量单金属及多金属MOFs材料被合成出来。用于制备MOFs材料的金属元素由过渡元素(Zn、Cu、Cd、Mn、Ni、Co、Cr、Fe、Ti、Zr等)拓展到主族元素(Al、In)和稀土元素(Tb、Sc、Sm、Dy、Ho、Er、Yb等)。另外,MOFs材料具有丰富多样的结构,骨架中含有大量金属源和碳源,为单金属氧化物(如ZnO、CuO、Cu2O、Mn2O3、Mn3O4、Cr2O3、Fe2O3、Al2O3、Co3O4、In2O3等)、多金属氧化物(如CuO/CeO2、CuO/TiO2、Fe2O3@TiO2、Cr2O3/Al2O3等)、金属氧化物/碳复合物(如Co3O4/N-doped NPC(NPC=nanoporous carbon)、ZnO/C)和金属/碳复合材料(如Co@C-N)等的制备提供了丰富的前驱体。在合适的条件下热解MOFs材料可得到结构丰富、性能优异的多孔金属氧化物和多孔金属氧化物/碳复合材料催化剂。
本文主要综述了以MOFs为前驱体的单金属氧化物、多金属氧化物、金属氧化物/碳复合材料和金属/碳复合材料等在催化方面的应用。
1 MOFs制备金属氧化物的方法
以MOFs为前驱体制备金属氧化物的方法主要是一步焙烧法和两步焙烧法,一步焙烧法是将MOFs直接在空气或惰性气氛中焙烧得到金属氧化物;两步焙烧法是先将MOFs在惰性气氛中焙烧,得到碳包覆的金属或金属氧化物,再在空气中焙烧得到金属氧化物。Cho等[37]将In-MOFs材料CPP-3和CPP-6在空气中焙烧得到两种结构的In2O3。多孔CPP-3在400~460 ℃快速热解为无孔In2O3;无孔CPP-6是在250~730 ℃缓慢热解为多孔In2O3。Xu等[38]以MIL-88-Fe为前驱体,分别采用两步焙烧法和一步焙烧法制备α-Fe2O3。先在惰性氛围中焙烧产生的碳有效地阻止金属氧化物的团聚,获得了孔径为~10nm的α-Fe2O3,且粒径更小,比表面积更大。Mahata等[39]以双金属MOFs材料[CoMn2{C6H3(COO)3}2]为前驱体得到了4种双金属氧化物CoMn2O4。CoMn2O4纳米粒子的尺寸可通过控制焙烧温度实现调控。Zhao等[40]以双金属MOF材料ZnMn2-ptcda(ptcda=perylene-3, 4, 9, 10-tetracarboxylic dianhydride)为前驱体制备了双金属氧化物ZnMn2O4。通过简单的热处理工艺,可以将双金属MOF前体的形貌精确转移到金属氧化物。
2 以MOFs为前驱体的金属氧化物催化剂
2.1 CO氧化
2.1.1 富氢气氛中CO优先氧化
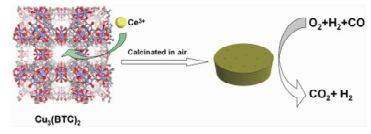
质子交换膜燃料电池被认为是电动汽车、中小型电站和移动设备等提供能源的最佳候选[41]。阳极是氢燃料发生氧化的场所,即2H2→4H++4e-。当燃料H2中含有少量CO时,CO在阳极Pt表面的强烈吸附作用会使阳极中毒,导致电池性能下降[42]。CO优先氧化是除去富氢氛围中CO的有效方法之一[43, 44]。CuO/CeO2是富氢气氛中CO优先氧化的良好催化剂[45]。Zhang等[46]先通过浸渍法把Ce3+引入Cu-BTC孔道,再利用一步焙烧法将Cu-BTC转化为CuO/CeO2。高焙烧温度会导致纳米粒子发生团聚,尺寸增加,BET比表面积下降,但有利于降低催化剂中C的含量。当焙烧温度达到700℃时,纳米粒子的尺寸急剧增加,而BET比表面积快速下降。600℃下获得的CuO/CeO2粒径均一,尺寸为15.3nm,BET比表面积最大,CuO纳米粒子高度分散在CeO2表面。Cu的含量对CuO/CeO2的催化性能有显著影响。随着CuO的含量的增加,CuO纳米粒子发生团聚,会削弱CuO与CeO2之间的相互作用,降低催化活性。5%-CuO/CeO2-600的催化活性最好,能在100℃下催化富氢气氛中CO完全转化为CO2(图 1)。
图 1
以Ce(NO3)3-Cu3(BTC)2为前驱体制备的CuO/CeO2催化CO氧化[46]
Figure 1.
CuO/CeO2 prepared from Ce(NO3)3-Cu3(BTC)2 precursor for preferential CO oxidation[46]
2.1.2 CO氧化
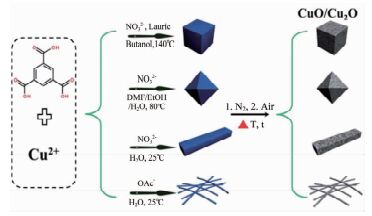
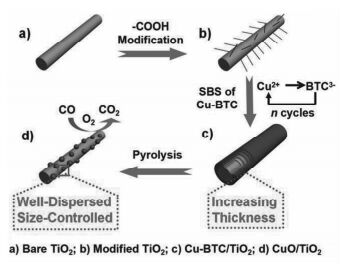
随着我国经济社会的快速发展,大气污染问题日益突出,对人们的健康产生了严重的威胁。CO是大气中一种常见的污染物。据报道,大气中80%的CO来自于汽车尾气的排放[47]。CO氧化能有效降低汽车尾气中CO污染物的排放,具有重要的现实意义。CuO是CO氧化催化剂中常见的活性成分[48~50]。Zhang等[51]利用两步焙烧法将Cu-BTC转化为介孔CuO/Cu2O复合物,平均孔径为4.4~13.1 nm,BET表面积为3.2~24.5 m2/g,复合物均保留了相应前驱体的形貌(图 2)。通过调控热解温度和时间可调节催化剂中CuO和Cu2O的含量。升高热解时的温度和延长时间会导致催化剂中活性组分Cu2O的含量下降,催化剂结构坍塌,形貌得不到保留。他们研究了4种CuO/Cu2O在CO氧化反应中的催化性能。结果表明,具有较大比表面积和较高Cu2O含量的棒状CuO/Cu2O的催化活性最好,八面体CuO/Cu2O的催化活性次之,线状CuO/Cu2O的催化活性最差。这是因为高比表面积有利于O2分子的吸附,为CO氧化反应提供更加充足的氧环境。在此基础上,刘朋飞等[52]将Au/Cu-BTC在体积比为2%的O2/He混合气中热解,得到了具有正八面体结构的介孔Au/CuxO催化剂。通过控制混合气中O2的浓度、热解时间调控催化剂中CuO和Cu2O的组成,获得了三种催化剂Au/Cu2O、Au/Cu2O-CuO和Au/CuO。随着热解时间的增加,催化剂中Cu2O的组成下降,CuO的组成增加。另外,热解时间对催化剂的比表面积和孔结构也有一定影响。延长热解时间,Au/CuxO催化剂的BET表面积和孔体积下降,孔径先增后降,同时Au纳米粒子发生团聚,尺寸增大,分散性下降。另外,Au的添加活化了Au纳米粒子晶界周围Cu2O中的晶格氧,产生氧空穴,提高了催化剂的活性。具有较大BET比表面积和较高Cu2O含量以及较好的Au纳米粒子分散性的Au/Cu2O在CO氧化反应中表现出优异的催化活性,可在90℃下催化CO氧化反应,180℃时CO转化完全。Liu等[53]在经均苯三甲酸改性后的TiO2上合成Cu-BTC,然后利用一步焙烧法将Cu-BTC/TiO2复合物在空气中转化为具有良好分散性和尺寸可控的CuO/TiO2催化剂(图 3)。CuO纳米粒子高度分散在TiO2表面,其尺寸随着Cu-BTC的厚度的增加而增加。当Cu-BTC的厚度小于33nm时,CuO纳米粒子的平均尺寸为约2nm。该催化剂在CO氧化反应中展现出优异的催化活性,能在175℃下将CO完全氧化为CO2。
2.2 催化产氢
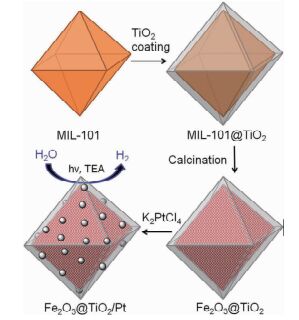
氢气是一种高效、清洁的可再生能源,每燃烧1kg氢气约产生121061kJ热量,是汽油的2.7倍、甲烷的2.4倍[54]。利用光催化技术分解水制取H2是新能源领域研究的热点之一[55]。TiO2是一种最常见的半导体光催化剂[56~58],但由于TiO2的光生电子与空穴易复合,且禁带宽度(3.2eV)大,故量子效率低,光谱响应范围窄,太阳光的利用率低[59]。半导体复合可降低光生电子-空穴对的复合,且能有效拓展半导体催化剂的光谱响应范围,是改善半导体催化剂的有效方法之一[60, 61]。李秀莹等[62]利用禁带宽度(2.2eV)较小的Fe2O3对TiO2进行改性,得到了Fe2O3-TiO2催化剂。结果表明,Fe2O3-TiO2的光谱响应范围拓展到了可见光区,太阳光的利用效率更高,且光生电子-空穴的复合率明显降低,在光催化降解亚甲基蓝时表现出优异的催化活性。Krafft等[63]通过在MIL-101(Fe)上先包覆TiO2再热解的方法得到了具有八面体核壳结构Fe2O3@TiO2,再通过还原四氯铂酸钾将贵金属Pt纳米粒子负载在Fe2O3@TiO2上,得到Fe2O3@TiO2/Pt(图 4)。Pt的费米能级较半导体的更低,光生电子易从半导体表面迁移至Pt纳米粒子上,Pt的引入有助于进一步提高光生电子-空穴对的分离,提高了量子效率。Fe2O3@TiO2/Pt催化剂可在450W氙灯照射下高效催化水分解制取H2。但该催化剂的稳定性较差,在循环使用3次后,部分八面体壳破裂。
CuO是一种P型半导体,禁带宽度为1.83~2.08 eV,对可见光有响应[64]。利用CuO对TiO2改性有利于光生电子的转移,降低光生电子-空穴的复合率,提高催化剂的光催化活性[65]。王萌等[66]先在羧基改性的A-B混合晶型介孔TiO2上合成前驱体Cu-BTC-TiO2,然后将其在空气中焙烧得到介孔CuO-TiO2,其平均孔径为7.35nm,BET比表面积为85.46m2/g。UV-Vis分析结果表明,与沉淀法得到的CuO-TiO2相比,CuO-TiO2的禁带宽度(2.95eV)更小,光谱响应范围更宽。SEM和HRTEM测试显示,TiO2呈纤维状,CuO纳米粒子均匀地生长在TiO2表面,粒径约为10nm。CuO纳米粒子在反应过程中不易团聚,稳定性也较沉淀法得到的CuO-TiO2好。以甲醇作牺牲剂,CuO-TiO2能在300W氙灯照射下高效催化水分解制取H2,平均产氢速率为0.32mmol/(g·h)。循环使用11次,催化活性基本保持不变,产氢速率也没有明显下降。
2.3 异丁烷脱氢
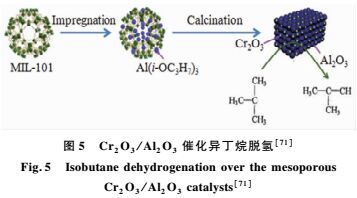
异丁烯是一种重要的基础化学品,特别是高纯度异丁烯更是稀缺。异丁烷脱氢为异丁烯的合成提供了一条新途径,也使C4产业链得到了很好的延伸[67]。异丁烷脱氢的催化剂主要有Pt系催化剂和Cr系催化剂。与Pt系催化剂相比,Cr系催化剂价格便宜,对原料中杂质含量要求较低,具有较好的发展前景[68]。其中,Cr2O3/Al2O3是一种具有较高催化活性和选择性的Cr系催化剂[69, 70]。Zhao等[71]先采用浸渍法将异丙醇铝负载在MIL-101(Cr)骨架上,再将其在空气中热解制得系列介孔Cr2O3/Al2O3,其孔尺寸为7.79~17.22 nm,BET比表面积为149.4~381.6 m2/g,孔体积为0.77~1.24 cm3/g。他们研究了该系列Cr2O3/Al2O3催化剂在异丁烷脱氢反应中的催化性能(图 5)。Cr3+是异丁烷脱氢反应的活性位,Cr3+/Cr6+的摩尔比对Cr2O3/Al2O3在异丁烷脱氢反应中的选择性具有重要影响。增加催化剂中Cr2O3含量和焙烧温度可提高催化剂中Cr3+的含量和Cr3+/Cr6+的摩尔比。不过,当Cr2O3的含量超过10(wt)%时,纳米粒子发生团聚,分散度下降,阻塞部分孔道,比表面积降低。载体Al2O3的表面酸性会使异丁烯被氧化生成CO2、CO和低链烷烃,造成积碳,导致催化剂的活性和选择性降低。他们还详细研究了助剂K2O对Cr2O3/Al2O3催化异丁烷脱氢反应的影响。结果显示,K2O的加入会降低催化剂的比表面积和表面Cr3+/Cr6+摩尔比,但可有效延长Cr2O3/Al2O3的寿命,极大提高催化剂的选择性和稳定性。K2O的负载量为1.5(wt)%,800℃下焙烧得到的K2O-Cr2O3/Al2O3能在600℃下催化异丁烷脱氢反应,异丁烯的选择性高达93.2%,异丁烷的转化率为60.1%。催化剂循环使用10次,异丁烯的选择性仍有92.7%。
2.4 环己烯氧化
环己烯分子中含有一个碳碳双键和多个活泼的α-H原子,通过对其进行选择性氧化可得到多种重要的化学品,如1, 2-环氧环己烷、环己烯酮和环己烯醇等[72, 73]。Mn2O3在NOx还原、CO氧化、氧还原反应、环己烷氧化等多个反应中表现出优异的催化活性[74~77]。Nayak等[78]将由氯化锰、丙三醇和叠氮化钠构筑的3D网络结构Mn-MOF(图 6(a))在空气中转化为具有海绵状结构的多孔α-Mn2O3(图 6(b)、6(c)),BET比表面积为52m2/g。他们对Mn-MOF热解生成α-Mn2O3的机理进行了详细研究。Mn-MOF中的结晶水在190℃下开始蒸发,并与骨架上N3-反应生成N2和H2,生成的气体穿过Mn-MOF表面,导致光滑的表面逐渐消失,并形成孔;随着温度的升高,Mn-MOF转化为无定形中间体;当温度达到500℃时,海绵状结构的α-Mn2O3生成。他们还研究了α-Mn2O3在环己烯氧化反应中的催化活性,氧化反应主要发生在烯丙基上,反应产物以2-环己烯-1-醇和2-环己烯酮为主。
2.5 醇直接氧化为酯
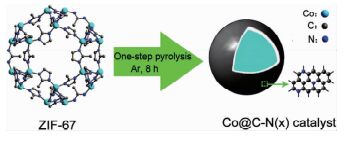
酯是一类重要的有机合成中间体和化工产品,在有机合成、香料、医药等多个领域具有重要的应用[79]。醇直接氧化生成酯避免使用羧酸或羧酸衍生物,是一条经济、绿色的合成路线。用于醇直接氧化制备酯的催化剂多为贵金属(如Pd、Au、Ru、Ir等)基催化剂,价格昂贵,且大多是均相催化剂,不易与产物分离[80, 81]。Jagadeesh等[82]报道的Co3O4-N@C多相催化剂在醇直接氧化制备酯反应中具有良好的催化活性,但反应中仍需添加碱性助剂K2CO3促进醇脱氢过程,反应温度较高。Zhong等[83]通过一步焙烧法将ZIF-67转化为富氮的纳米复合材料Co@C-N(图 7),Co含量约为30(wt)%~40(wt)%,BET比表面积为300~400 m2/g,孔径约为5.09~6.11 Å。ZIF-67是一种富氮无氧的Co基MOF材料,稳定性较好,惰性气氛中缓慢碳化最大限度地保留前驱体中N元素和C元素,这两种元素的存在有效阻止了Co的烧结和团聚,提高了Co纳米粒子的分散性。焙烧温度对Co@C-N的BET比表面积和孔结构具有重要影响。随着焙烧温度的升高,Co@C-N的BET比表面积呈现先增加后下降的趋势,700℃下的BET比表面积最大,孔径逐渐增加,当温度增加至900℃时,Co纳米粒子发生严重团聚,尺寸增加,BET比表面积下降。他们研究了不同焙烧温度下得到的Co@C-N对芳香醇与脂肪醇氧化交叉酯化反应的催化性能,结果表明,Co@C-N(800℃)的催化活性最好,能在室温、无碱条件下高效催化芳香醇与脂肪醇氧化交叉酯化反应,相应酯的收率为80%至>99%,反应条件温和。这是因为800℃焙烧制备的Co@C-N中N的含量高于700℃焙烧的样品,而N元素有利于电子的活化和迁移,促进反应的进行。苯环上取代基类型和位置对反应有明显的影响,吸电子基团如-CO2CH3、-NO2、-F、-Cl、-Br和-I显示出更好的反应活性,相应酯的收率大于99%;取代基在对位的反应活性大于取代基在邻位和间位。随着脂肪醇碳链长度的增长,相应酯的收率下降;直链脂肪醇的反应活性大于有支链的脂肪醇。
Zhou等[84]在N2氛围中不同温度下将ZIF-67转化为介孔Co-CoO@N-doped NPC复合物(图 8)。Co-CoO@N-doped NPC保留了前驱体的形貌,只是有些许凹陷,尺寸较前驱体稍微收缩,5~20 nm的Co和CoO纳米粒子均匀地分散在多孔碳基底上。这种多孔结构有效抑制了活性纳米粒子团聚,有利于反应物分子和产物分子的扩散。随着焙烧温度的增加,Co-CoO@N-doped NPC中Co含量增加,金属Co的结晶度提高,BET表面积先增后减。700℃下得到的纳米复合物BET比表面最大,孔径为2.5nm。其在芳香醇与甲醇制备酯的反应中表现出良好的催化活性,具有较高的反应物转化率和产物选择性,但反应过程中需添加碱性助剂K2CO3。这种纳米复合物还具有强磁性,在外加磁场的作用下,Co-CoO@N-doped NPC很容易从反应溶液中分离回收。
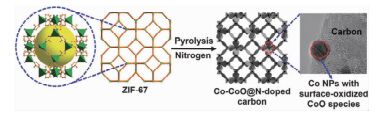 图 8
ZIF-67热解制备Co-CoO@N掺杂多孔碳纳米复合材料[84]
Figure 8.
Synthesis of Co-CoO@N-doped porous carbon nanocomposites via the pyrolysis of ZIF-67
图 8
ZIF-67热解制备Co-CoO@N掺杂多孔碳纳米复合材料[84]
Figure 8.
Synthesis of Co-CoO@N-doped porous carbon nanocomposites via the pyrolysis of ZIF-67
2.6 醛氧化酰胺化反应
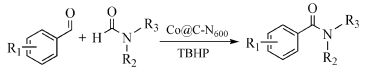
醛氧化酰胺化反应是合成含有酰胺键的天然产物、材料以及药物分子中一类非常重要的化学反应[85]。Bai等[86]将由2, 4, 6-三(4-吡啶)-1, 3, 5-三嗪和均苯三酸混合配体构筑的无孔Co-MOF材料在氩气中焙烧得到多孔材料Co@C-N,Co的含量为30%~45%,并研究了它们在苯甲醛及其衍生物与甲酰胺衍生物氧化酰胺化反应中的催化性能(图式1)。焙烧温度对Co@C-N的组成、BET表面积和孔结构具有重要影响。随着焙烧温度的提高,Co@C-N中C、N的含量逐渐下降,Co的含量逐渐增加;焙烧产物的BET比表面积、孔体积较前驱体都有大幅增加,呈现出先增后降的变化趋势。600℃下得到的Co@C-N的BET比表面积(251m2/g)最大,孔径(4.9Å)最小,孔体积为0.11cm3/g,C、N和Co的质量百分比分别是61%、1.5%和35.8%;Co纳米粒子的尺寸为7nm,高度分散在N掺杂的多孔碳基底上。催化剂在苯甲醛及其衍生物与甲酰胺衍生物氧化酰胺化反应中的催化活性最好,酰胺的收率达到约90%。另外,反应物结构对酰胺化反应有一定影响,富电子芳香醛的反应活性高于缺电子芳香醛的反应活性;具有长碳链烷基甲酰胺的反应活性较低。
2.7 光催化降解有机物
利用光催化技术降解水体中的有机污染物被认为是一项具有发展前景的污水处理技术[87]。Mn3O4被广泛用于去除废气中的有害气体(如CO、NOx等)和有机物[88]。Peng等[89]将Mn-MOF材料在空气中转化为介孔Mn3O4,BET比表面积(160.4m2/g)和孔体积(0.649cm3/g)远远高于由MnOOH(32.88m2/g,0.053cm3/g)[90]和Mn2O3(99.7m2/g)[91]得到的Mn3O4;其孔尺寸为23nm,也高于已报道的介孔Mn3O4(3~4 nm)[90, 91]。前驱体和焙烧产物Mn3O4的孔尺寸可通过改变Mn-MOF合成时模板剂的组成实现简单调控。他们将Mn3O4用作光催化降解亚甲基蓝的催化剂。结果显示,该介孔Mn3O4对亚甲基蓝具有很高的催化降解效率,光照1.5h,降解率超过99.7%,催化活性高于文献报道的Mn3O4[92]。
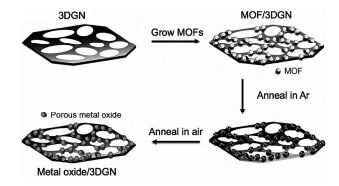
ZnO具有一定的光催化活性,且无毒、成本低,是近年来研究较多的半导体光催化剂之一[93]。三维石墨烯(3DGN)是一种新型的功能材料,具有多孔性、比表面积大、机械强度高、电子传导能力强等优点,可用作催化剂和催化剂载体[94]。Cao等[95]先在3DGN上合成ZIF-8,再利用两步焙烧法将ZIF-8/3DGN复合物转化为ZnO/3DGNs复合物(图 9)。在氩气中焙烧时,ZIF-8的有机配体转化生成无定形碳,包覆在纳米粒子(尺寸约为10nm)表面,有效防止了纳米粒子的团聚;空气中焙烧后,无定形碳发生分解,纳米粒子的尺寸变小。另外,前驱体ZIF-8和ZnO纳米粒子的尺寸可通过控制MOF的合成条件进行调控。他们研究了ZnO/3DGNs复合物在光催化降解亚甲基蓝反应中的催化活性。结果显示,ZnO/3DGNs复合物的光催化活性明显优于3DGNs和ZnO,紫外光照射60min,亚甲基蓝被完全降解。这是因为3DGN与ZnO之间良好的协同作用使得光生电子-空穴对得到及时分离,提高了量子利用效率。另外,ZnO/3DGNs还具有良好的机械性能,可直接用镊子把ZnO/3DGNs从反应液中取出,实现催化剂与反应液的简单分离,用超纯水洗涤干净就可再次使用。重复使用4次后ZnO/3DGNs的光催化活性基本保持不变,表现出良好的循环使用性能。郭艳蓉等[96]将MOF-5直接在氮气中焙烧得到ZnO/C催化剂,其BET比表面积高达390m2/g,孔体积为0.599mL/g。惰性气氛中高温处理有效保留了前驱体的高比表面积和多孔结构,且高温产生的非晶态多孔碳也有效阻止了ZnO纳米粒子团聚。负载银后催化剂的光谱响应范围扩展到了可见光区,提高了太阳光的利用率,Ag/ZnO/C的BET比表面积略有下降,为232m2/g,Ag纳米粒子的平均尺寸为30nm。他们研究ZnO/C和Ag/ZnO/C光催化降解亚甲基蓝的催化性能,结果显示,ZnO/C和Ag/ZnO/C对亚甲基蓝都具有较高的降解效率。高温处理产生的非晶态的碳对原位生成的ZnO有很好的保护作用,减缓了ZnO的光腐蚀,同时非晶态碳提供了很多电子陷阱,使光生电子易流向非晶态碳表面,抑制了光生电子-空穴对的复合。负载Ag后,光生电子流向了费米能级较低的Ag纳米粒子表面,进一步抑制了光生电子-空穴对的复合,提高催化剂的稳定性和催化活性。Ag/ZnO/C在循环使用5次后,降解率几乎保持不变;而ZnO/C在第3次使用时,光催化性能明显下降。
CuO也是一种常见的半导体光催化剂[65, 66]。Fan等[97]先在水热条件下合成了两种形貌不同的Cu-MOFs前驱体:2D结构的蝴蝶状Cu(C6H4NO2)2(H2O)4纳米片和1D结构的[Cu(C6H4NO2)(OH)]H2O纳米棒,再将它们分别在空气中下热处理得到的CuO。2D结构的蝴蝶状Cu(C6H4NO2)2(H2O)4纳米片转化为蝴蝶状CuO纳米片,由20~50 nm的纳米粒子组成;[Cu(C6H4NO2)(OH)]H2O纳米棒转化为CuO纳米管,由许多具有三角形截面的一维纳米结构组成。与蝴蝶状CuO纳米片相比,CuO纳米管具有更高的BET比表面积,在光催化降解若丹明B反应中表现出较好的光催化活性,可见光照射40min,若丹明B的降解率达到了92%。
2.8 氧还原反应
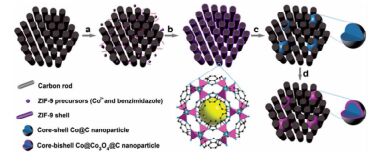
氧还原反应(ORR)是燃料电池中重要的阴极反应,该反应较阳极的氧化反应慢得多,需用价格昂贵的Pt及其合金作为催化剂,高昂的成本限制了燃料电池的商业化发展。研究表明,碳负载金属氧化物,特别是钴基氧化物是碱性介质中ORR反应的良好催化剂,金属氧化物与载体碳之间的耦合作用能明显提高催化剂的ORR活性[98, 99]。Chaikittisilp等[100]将ZIF-9在N2惰性气氛中碳化得到Co/N-doped NPC,再将碳化产物在空气中氧化得到Co3O4/N-doped NPC,其主要为微孔和介孔结构,BET比表面积为75~280 m2/g,孔体积为0.34~0.45 cm3/g。实验结果表明,高温碳化有利于生成更稳定的多孔碳网络结构,而高温氧化会导致生成的氧化钴纳米粒子发生团聚,破坏多孔碳网络结构,使比表面积下降;但在低温下,只有极小部分金属被氧化。另外,多孔碳中掺杂的N增强了多孔碳网络结构与活性组分Co3O4之间的耦合作用,提高了Co3O4/N-doped NPC在氧还原反应中的催化活性。800℃下碳化、300℃下氧化得到Co3O4/N-doped NPC的ORR活性最好,其电子转移数接近理论值,达到了3.6,高于商业Pt/CB催化剂[101]。此外,Co3O4/N-doped NPC还具有优良的析氧反应催化活性,是一种双功能催化剂。Zhang等[102]将Co-MOF材料[Co3(μ2-OH)4(I)2]·2H2O(I为次黄嘌呤)在N2中一步碳化得到核壳结构Co3O4@N-doped NPC。核(Co3O4纳米粒子)与壳(N-doped NPC)之间的空间可看作一个纳米反应器。这种核壳结构提高了Co3O4纳米粒子的ORR催化活性。Co3O4@N-doped NPC的ORR起始还原电位和峰电位分别为0.95和0.8 V,接近商业Pt/C的起始还原电位和峰电位;电子转移数高达为3.93,与商业Pt/C相当;同时,Co3O4@N-doped NPC还具有很好的甲醇耐受性。同年,Xia等[103]先在硝酸改性的多孔碳(CM)上合成ZIF-9,再利用两步焙烧法得到核-双壳结构Co@Co3O4@C纳米粒子,Co@Co3O4@C纳米粒子高分散在多孔碳中(图 10)。Co@Co3O4@C-CM为高度有序的介孔材料,BET比表面积高达610m2/g。这种核-双壳结构增强了金属氧化物与碳壳之间的相互作用,提高了纳米粒子与多孔碳之间的电子转移和相互作用,使得纳米粒子不易从多孔碳基底上脱落。此外,多孔碳提供了一个三维互联的孔隙结构,有利于Co@Co3O4@C纳米粒子充分暴露在氧气中。Co@Co3O4@C-CM的ORR反应催化活性明显高于Co3O4/N-doped NPC[100]。
3 结语
综上所述,以MOFs为前驱体的多孔金属氧化物和多孔金属氧化物/碳复合物已成功用作CO氧化、催化产氢、异丁烷脱氢、环己烯氧化、醇直接氧化为酯、醛氧化酰胺化反应、光催化降解有机物、氧还原反应等多个反应的催化剂,并展现出良好的催化活性,这些研究工作极大地拓宽了MOFs材料的应用范围。MOFs作为前驱体制备多孔金属氧化物具有以下五点优势:(1) MOFs材料由金属离子和有机配体组成,大量的金属离子和无数的有机配体可以构筑出结构多样、种类繁多的MOFs,混合配体的使用更加增添了MOFs种类的多样性,为多种金属氧化物、金属氧化物/碳复合材料、金属/碳复合材料等功能材料的制备提供了丰富的前驱体;(2) MOFs骨架中高度有序的金属离子被有机配体很好地分离开,这一特性有利于解决焙烧过程中因金属烧结而出现纳米粒子团聚这一问题,可制备出高分散性金属氧化物催化剂;(3) 制备方法简单,预见性好,焙烧产物形态均一,且可控性也较强,尤其是制备核壳结构金属氧化物时较传统的液相沉积法更加简便;(4) MOFs的无机金属节点和有机配体易功能化,通过向骨架引入功能性金属节点或官能团进行修饰,可获得高性能MOFs前驱体,间接实现对金属氧化物和金属氧化物/碳复合物的化学组成、物理化学性质的调控,为获得高性能多金属氧化物及其复合物和金属氧化物/碳复合物提供了可能;(5) MOFs易与其他材料(如三维石墨烯、多孔碳等)复合,制备出性能优异的复合物前驱体,为获得具有特殊形貌、结构的金属氧化物/碳复合物提供了可能。
-
-
[1]
Y Liu, J Deng, S Xie et al. Chin. J. Catal., 2016, 37(8):1193~1205.
-
[2]
M Abirami, S M Hwang, J Yang et al. ACS Appl. Mater. Interf., 2016, 8(48):32778~32787.
-
[3]
T Graunke, K Schmitt, J W llenstein. Sensors, 2016, 2016(24):1~22.
-
[4]
D Zhang, J Liu, B Xia. J. Electron. Mater., 2016, 45(8):4324~4330.
-
[5]
L Q Qwabe, V D B C Dasireddy, S Singh et al. Int. J. Hydrogen Energy, 2016, 41(4):2144~2153.
-
[6]
N R Elezovic, V R Radmilovic, N V Krstajic. RSC Adv., 2016, 6(8):6788~6801.
-
[7]
J J H B Sattler, J Ruiz~Martinez, E Santillan~Jimenez et al. Chem. Rev., 2014, 114(20):10613~10653.
-
[8]
李军, 潘磊, 王际童等. 无机材料学报, 2016, 31(11):1205~1211.
-
[9]
J Liu, S Zou, L Xiao et al. Catal. Sci. Technol., 2014, 4(2):441~446.
-
[10]
Z Haider, Y S Kang. ACS Appl. Mater. Interf., 2014, 6(13):10342~10352.
-
[11]
Z Wu, M Li, S H Overbury. J. Catal., 2012, 285(1):61~73.
-
[12]
邓积光, 何胜男, 谢少华等. 高等学校化学学报, 2014, 35(6):1119~1129.
-
[13]
J Li, C Ma, X Xu et al. Environ. Sci. Technol., 2008, 42(23):8947~8951.
-
[14]
C Y Ma, Z Mu, J J Li et al. J. Am. Chem. Soc., 2010, 132(8):2608~2613.
-
[15]
陈英. 中国科学院大学硕士学位论文, 2015.
-
[16]
X Chen, T Yu, X Fan et al. Appl. Surf. Sci., 2007, 253(20):8500~8506.
-
[17]
J Y Luo, M Meng, X Li et al. J. Catal., 2008,254(2):310~324.
-
[18]
G A Seisenbaeva, M P Moloney, R Tekoriute et al. Langmuir, 2010, 26(12):9809~9817.
-
[19]
S W Liu, C Li, J G Yu et al. Cryst. Eng. Commun., 2011, 13(7):2533~2541.
-
[20]
A Sinhamahapatra, A K Giri, P Pal et al. J. Mater. Chem., 2012, 22(33):17227~17235.
-
[21]
王丽苹, 王公应. 分子催化, 2015, 29(3):275~288.
-
[22]
H Li, M Eddaoudi, M O'Keeffe et al. Nature, 1999, 402(6759):276~279.
-
[23]
M Eddaoudi, K Jaheon, R Nathaniel et al. Nature, 2002,295(5554):469~472.
-
[24]
J Albero, H García. New Mater. Catal. Appl., 2016, 23(3):13~40.
-
[25]
A Arnanz, M Pintado-Sierra, A Corma et al. Adv. Synth. Catal., 2012, 354(7):1347~1355.
-
[26]
N T S Phan, K K A Le, T D Phan. Appl. Catal. A-Gen., 2010, 382(2):246~253.
-
[27]
I A Khan, Y Qian, A Badshah et al. ACS Appl. Mater. Interf., 2016, 8(27):1768~17275.
-
[28]
M Sabo, A Henschel, H Fr de et al. J. Mater. Chem., 2007, 17(36):3827~3832.
-
[29]
S Gao, Z Nan, M Shu et al. Appl. Catal. A-Gen., 2010, 388(1):196~201.
-
[30]
Y X Zhou. Aata Phys-Chem Sin., 2010, 26(4):939~945(7).
-
[31]
L P Wang, B Xiao, G Y Wang et al. Sci. China:Chem., 2011, 54(9):1468~1473.
-
[32]
L Wang, G Wang, F Wang et al. Asian J. Chem., 2013, 25(10):5385~5389.
-
[33]
王丽苹, 王公应, 汪帆等. 高分子材料科学与工程, 2013(3):82~85.
-
[34]
L Alaerts, E Séguin, H Poelman et al. Chem. Eur. J., 2006, 12(28):7353~7363.
-
[35]
Y Zhao, C Zhong, C J Liu. Catal. Commun., 2013, 38(5):74~76.
-
[36]
W A Qiu, Y Wang, L I Chuanqiang et al. Chin. J. Catal., 2012, 33(s 4/6):986~992.
-
[37]
W Cho, Y H Lee, H Lee J et al. Chem. Commun., 2009, (31):4756~4758
-
[38]
X D Xu, R G Cao, S Y Jeong et al. Nano Lett., 2012, 12(9):4988~4991.
-
[39]
P Mahata, D Sarma, C Madhu et al. Dalton Transac., 2011, 40(9):1952~1960.
-
[40]
J Zhao, F Q Wang, P P Su et al. J. Mater. Chem., 2012, 22(26):13328~13333.
-
[41]
汪嘉澍, 潘国顺, 郭丹. 化学进展, 2012, 24(10):1906~1914.
-
[42]
B T Qiao, J X Liu, Y G Wang et al. ACS Catal., 2015, 5(11):6249~6254.
-
[43]
M F Luo, J M Ma, J Q Lu et al. J. Catal., 2006, 246(1):52~59.
-
[44]
G Avgouropoulos, T Ioannides. Appl. Catal. B-Environ., 2006, 67(1):1~11.
-
[45]
士丽敏, 郑德山, 王素敏. 化工进展, 2011, 30(9):1956~1960.
-
[46]
F Zhang, C Chen, W M Xiao et al. Catal. Commun., 2012,26(35):25~59.
-
[47]
陈庆锋,付英, 卢艳等. 环境污染与健康. 化学工业出版社, 2014:16.
-
[48]
M F Luo, J M Ma, J Q Lu et al. J. Catal., 2007, 246(1):52~59.
-
[49]
Y Feng, X Zheng. Nano Lett., 2010, 10(11):4762~4766.
-
[50]
戴越, 李珊珊, 汤常金等. 无机化学学报, 2012, 28(8):1555~1562.
-
[51]
S Y Zhang, H Liu, C C Sun et al. J. Mater. Chem. A, 2015, 3(10):5294~5298.
-
[52]
刘朋飞, 张所瀛, 杨祝红等. 化工学报, 2016, 67(6):2325~2331.
-
[53]
H Liu, S Y Zhang, Y Y Liu et al. Small, 2015, 11(26):3130~3134.
-
[54]
张轲, 刘述丽, 刘明明等. 材料导报, 2011, 25(9):116~119.
-
[55]
温福宇, 杨金辉, 宗旭等. 化学进展, 2009, 21(11):2285~2302.
-
[56]
Z W Shi, M Guo, L J Wang et al. Chin. J. Chem. Phys., 2016(2):199~204.
-
[57]
罗利军, 王娟, 潘学军等. 化学通报, 2013, 76(4):332~337.
-
[58]
Y Lu, Y P Zang, H M Zhang et al. Sci. Bull., 2016, 61(13):1~10.
-
[59]
D Li, H Haneda, A Shunichi-Hishita et al. Chem. Mater., 2005, 17(10):2596~2602.
-
[60]
B Palanisamy, C M Babu, B Sundaravel et al. J. Hazard. Mater., 2013, 252~253C(4):233~242.
-
[61]
P Khemthong, P Photai, N Grisdanurak. Int. J. Hydrogen Energ., 2013, 38(36):15992~16001.
-
[62]
李秀莹, 王靖宇, 王晓宇等. 高等学校化学学报, 2010, 31(4):662~666.
-
[63]
K E Krafft, C Wang, W B Lin. Adv. Mater., 2012, 24(15):2014~2018.
-
[64]
J Jun, C Jin, H Kim et al. Appl. Surf. Sci., 2009, 255(20):8544~8550.
-
[65]
杨旭, 李小龙, 胡彩花等. 无机化学学报, 2015, 31(11):2167~2173.
-
[66]
王萌,徐律,周燕南等. 南京工业大学学报(自然科学版), 2016, 38(2):88~93.
-
[67]
刘莹. 石油化工, 2016, 45(5):630~635.
-
[68]
谭晓林, 马波, 张喜文等. 化工进展, 2010, 29(1):51~57.
-
[69]
L L Xu, Z L Wang, H L Song et al. Catal. Commum., 2013, 35(17):76~81.
-
[70]
樊彤彤, 周广林, 房鑫等. 燃料化学学报, 2016, 44(9):1125~1130.
-
[71]
H H Zhao, H L Song, L L Xua et al. Appl. Catal. A-Gen., 2013,456(6):188~196.
-
[72]
W Nam, S Y Oh, J Kim et al. J. Org. Chem., 2003,68(20):7903~7906.
-
[73]
S Rayati, N Torabi, A Ghaemi et al. Inorg. Chim. Acta, 2008, 361(5):1239~1245.
-
[74]
M J Jeon, S H Park, J M Kim et al. J. Nanosci. Nanotechnol., 2014, 14(3):2527~2531.
-
[75]
H Q Dong, Y Y Chen, M Han et al. J. Mater. Chem. A, 2014, 2(5):1272~1276.
-
[76]
M Wu, W Zhan, Y Guo et al. Chin. J. Catal., 2016, 37(1):184~192.
-
[77]
R Dong, H Wang, Q Zhang et al. Cryst. Eng. Commun., 2015, 17(38):7406~7413.
-
[78]
S Nayak, S Malik, S Indris et al. Chem. Eur. J., 2010, 16(4):1158~1162.
-
[79]
石先莹, 韩晓燕, 马文娟等. 有机化学, 2011, 31(3):297~305.
-
[80]
T Nobuta, A Fujiya, S Hirashima et al. Tetrahed. Lett., 2012, 53(39):5306~5308.
-
[81]
X F Wu. Chem. Eur. J., 2012, 18(29):8912~8915.
-
[82]
R V Jagadeesh, H Junge, M M Pohl et al. J. Am. Chem. Soc., 2013, 135(29):10776~10782
-
[83]
W Zhong, H L Liu, C H Bai et al. ACS Catal., 2015, 5(3):1850~1856
-
[84]
Y X Zhou, Y Z Chen, L N Cao et al. Chem. Commun., 2015, 51(39):8292~8295.
-
[85]
S C Ghosh, J S Y Ngiam, A M Seayad et al. J. Org. Chem., 2012, 77(18):8007~8015.
-
[86]
C H Bai, X F Yao, Y W Li. ACS Catal., 2015, 5(2):884~891.
-
[87]
刘芳, 樊丰涛, 吕玉翠等. 化工学报, 2016, 5(5):1635~1643.
-
[88]
唐爱东, 黄可龙. 无机化学学报, 2005, 21(6):929~932.
-
[89]
L Peng, J L Zhang, Z M Xue et al. Chem. Commun., 2013,(49):11695~11697.
-
[90]
Z C Bai, B Sun, N Fan et al. Chem. Eur. J., 2012,18(25):5319~5324.
-
[91]
T Yousefi, A N Golikand, M H Mashhaddizadeh et al. Curr. Appl. Phys., 2012,12(10):544~549.
-
[92]
P Q Zhang, Y G Zhan, B G Cai et al. Nano Res., 2010, 3(4):235~243.
-
[93]
W Yu, J Zhang, T Peng. Appl. Catal. B-Environ., 2016, 181(12):220~227.
-
[94]
周国珺, 叶志凯, 石微微等. 化学进展, 2014(6):950~960.
-
[95]
X H Cao, B Zheng, X H Rui et al. Angew. Chem. Int. Ed., 2014, 53(5):1404~1409.
-
[96]
郭艳蓉, 常薇, 张雯等. 无机材料学报, 2015, 30(12):1321~1326.
-
[97]
Y Z Fan, R M Liu, W Du et al. J. Mater. Chem., 2012, 22(22):12609~12617.
-
[98]
Y Tan, C Xu, G Chen et al. Adv. Funct. Mater., 2012, 22(20):4584~4591.
-
[99]
Z S Wu, S Yang, Y Sun et al. J. Am. Chem. Soc., 2012, 134(22):9082~9085.
-
[100]
C Chaikittisilp, N L Torad, C L Li et al. Chem. Eur. J., 2014, 20(15):4217~4221.
-
[101]
Y Y Liang, Y G Li, H L Wang et al. J. Am. Chem. Soc., 2013, 135(6):2013~2036.
-
[102]
G J Zhang, C X Li, J Liu et al. J. Mater. Chem. A, 2014, 2(22), 8184~8189.
-
[103]
W Xia, R Q Zou, L An et al. Energ. Environ. Sci., 2015, 8(2):568~576.
-
[1]
-






图 8 ZIF-67热解制备Co-CoO@N掺杂多孔碳纳米复合材料[84]
Figure 8 Synthesis of Co-CoO@N-doped porous carbon nanocomposites via the pyrolysis of ZIF-67
-



 下载:
下载:










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 1173
- HTML全文浏览量: 271
 下载:
下载:
