 图 图式1
配合物42~50的制备
Figure 图式1.
Preparation of complexes 42~50
图 图式1
配合物42~50的制备
Figure 图式1.
Preparation of complexes 42~50
引用本文:
赵宁. 手性金属有机化合物催化丙交酯聚合反应研究进展[J]. 有机化学,
2017, 37(5): 1139-1159.
doi:
10.6023/cjoc201612011

Citation: Zhao Ning. Progress in Ring Opening Polymerization of Lactides Catalyzed by Chiral Organometallic Complexes[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1139-1159. doi: 10.6023/cjoc201612011
Citation: Zhao Ning. Progress in Ring Opening Polymerization of Lactides Catalyzed by Chiral Organometallic Complexes[J]. Chinese Journal of Organic Chemistry, 2017, 37(5): 1139-1159. doi: 10.6023/cjoc201612011
手性金属有机化合物催化丙交酯聚合反应研究进展
English
Progress in Ring Opening Polymerization of Lactides Catalyzed by Chiral Organometallic Complexes
Abstract:
Polylactides have received considerable attention in recent years due to their outstanding properties such as biocompatibility, biodegradability, and renewability. In particular, the studies on ring opening polymerization (ROP) of lactides in stereoselective manner catalyzed by chiral organometallic complexes have been regarded as one of the most significant fields in the synthesis of polylactides. The important advances in the ROP of lactides catalyzed by chiral organometallic complexes are reviewed in this article.
-
Key words:
- chiral organometallic complexes
- / catalysis
- / polymerization of lactides
-
生物降解材料, 聚丙交酯, 近年来在组织工程、药物缓释、环境材料、服装纤维、微电子等领域获得了广泛的应用[1~2].由于丙交酯有两个手性中心, 因此它具有四种存在形式, 即左旋丙交酯(L-LA)、右旋丙交酯(D-LA)、内消旋丙交酯(meso-LA)和外消旋丙交酯(rac-LA) (等物质的量的L-LA和D-LA的混合物).这四种形式的丙交酯在聚合过程中可以形成不同立构规整度的聚合物, 如等规、杂规、间规、无规或立体嵌段等(Eqs. 1~5)[3].聚丙交酯的立构规整度在很大程度上影响着它的物理性能和加工性能, 进而也决定了它的用途[1~2].因此, 近年来, 金属有机化合物在丙交酯立体选择性开环聚合反应中的应用研究引起了人们的广泛关注[4].其中, 利用手性金属有机化合物来催化丙交酯的立体选择性开环聚合反应成为该研究的热点之一.本文将对前人在手性金属有机化合物催化丙交酯开环聚合反应方面所取得的研究结果进行总结.
1 手性有机锌化合物催化的丙交酯聚合反应
由于聚丙交酯被广泛地应用于食品包装和生物医学等领域, 因此, 选择一种毒副作用小, 且生物相容的金属化合物作为催化剂备受科学家的青睐.进而, 手性锌催化剂的设计与合成得到了推崇.
2000年, Chisholm等[5]利用手性铊化合物1合成了手性锌化合物2 (Eq. 6), 并将它用于催化外消旋丙交酯(rac-LA)和内消旋丙交酯(meso-LA)的共聚反应.在聚合反应初期, 也就是单体转化率为30%以内时, 二者的聚合反应速度基本相同, 但之后, 内消旋丙交酯优先发生了聚合, 该催化剂显示了一定的非对映选择性.另外, 在催化外消旋丙交酯聚合反应时, 他们对反应液进行了旋光值的跟踪检测, 发现该手性锌催化剂对左旋丙交酯(L-LA)的开环聚合反应具有中等的对映选择性, 但作者未报道所得聚合物的规整度.
2009年, Mehrkhodavandi等[6]利用手性环己二胺衍生的配体3合成了相应的手性锌化合物4 (Eq. 7), 并将它用来催化外消旋丙交酯的聚合反应, 得到的是分子量比理论值大且分子量分布较宽的无规聚合物.动力学研究发现, 该催化剂在聚合过程中经历了3 h的链引发期.因此, 所得聚合物分子量的偏差和较宽的分子量分布可能是由于不完全引发造成的, 但造成该催化剂较长引发期的原因尚不清楚.
2010年, Schaper等[7]利用手性β-二酮亚胺化合物5合成了相应的异丙氧基锌化合物6 (Eq. 8), 并将其用来催化外消旋丙交酯的聚合反应.发现, 该催化剂在丙交酯聚合过程中具有较好的催化活性, 且生成了杂规聚合物, Pr值最高可达0.87.当转化率达到75%时, 剩余单体的ee值几乎为0.该手性催化剂在高分子链增长过程中没有发挥对映选择性的作用, 且链增长机理为链端控制机理.这是由于分子中N—C键容易发生旋转, 也就是配体刚性较弱, 因此它主要通过链端控制来生成杂规聚合物.
同年, Breuning等[8]将二胺基配体7分别与ZnCl2、Zn(OAc)2反应, 生成了相应的手性氯化锌络合物8和手性醋酸锌络合物9 (Eq. 9).随后, 他们考查了这两个络合物催化外消旋丙交酯的本体聚合反应(丙交酯熔融状态下).发现, 催化剂8没有催化活性, 而9在150 ℃下反应48 h可得到单体转化率为29%的聚合物, 但所得聚合物为无规聚合物, 说明催化剂9中的手性环境并不能控制反应的选择性.
与此同时, Darensbourg等[9]将天然氨基酸衍生的一系列手性Schiff碱配体10~12与Zn[N(SiMe3)2]2反应, 得到了相应的手性锌化合物13~15 (Eq. 10), 并进一步将这些化合物用于催化丙交酯的聚合反应.发现, 这些催化剂在室温下都显示了很好的催化活性, 生成分子量分布较窄的聚合物.随后, 他们将这些催化剂分别用于催化左旋和右旋丙交酯的聚合反应, 发现二者的反应速率相近(kD(obsd)/kL(obsd)≈1), 也就说这些手性催化剂没有显示出对映体选择性.但配体上的取代基和反应温度对选择性有较大影响.例如, -30 ℃下, 15催化外消旋丙交酯聚合反应可得到杂规聚合物, 且Pr值最高可达0.89.同时, 他们将不同规整度的杂规聚合物通过差示扫描量热仪(DSC)测定, 发现随着杂规度的提高, 玻璃化温度Tg值也逐渐升高, 说明聚合物的立构规整度决定了它的热稳定性.
2011年, Schaper等[10]将手性苯乙胺衍生的配体16和18分别与ZnEt2或Zn[N(SiMe3)2]2反应生成了相应的手性锌化合物17和19 (Eqs. 11, 12).随后, 他们将化合物17和19用来催化外消旋丙交酯的聚合反应.发现, 以CDCl3作溶剂时, 室温下17和19均无催化活性; 当反应温度升至50 ℃时, 2 h后化合物17催化外消旋丙交酯单体获得91%的转化率, 但聚合物的规整度不高(Pr=0.59);而催化剂19在50 ℃下依然没有催化活性, 当反应温度升至180 ℃催化本体聚合反应时, 反应0.5 h转化率可达95%, 所得聚合物近似无规(Pr=0.52).该反应温度下, 催化剂19可能发生了分解, 它的低催化活性, 可能是由于空间位阻造成的.
2012年, Otero等[11]将三齿配体20与Zn(CH2SiMe3)2反应生成了化合物21 (Eq. 13), 并将其用来分别催化左旋和外消旋丙交酯的聚合反应.发现, 在不加助催化剂的情况下, 50 ℃下可获得较高的转化率, 所得聚合物的分子量分布较窄且转化率与数均分子量成线性关系.进一步通过氢谱和MALDI-TOF质谱分析发现, 该聚合反应是通过烷基单点引发的.该催化剂催化外消旋丙交酯可得到杂规聚合物, Pr值最高可达0.77, 且随着反应温度的提高, Pr值降低.
同年, 我们[12a]将手性氮杂环丁烷配体22与ZnEt2反应生成了一个四核手性锌化合物23 (Eq. 14), 并进一步考查了它在外消旋丙交酯聚合反应中的应用.发现, 室温下该化合物可有效地催化丙交酯的聚合反应且催化活性较高, 所得聚合物的分子量分布较窄, 且为中等程度的杂规聚合物.另外, 还发现, 该催化剂的催化效果受溶剂的影响很大.例如, 在CH2Cl2中反应较慢, 且所得聚合物的分子量分布较宽.这可能是由于CH2Cl2与金属中心发生了配位作用引起的.
随后, 我们[12b]将联萘酚胺和联苯酚胺衍生的手性配体24~26与ZnEt2反应, 生成了三核手性锌化合物27~29 (Eqs. 15~17).发现, 这三个手性锌化合物都可有效地催化外消旋丙交酯的聚合反应, 所得聚合物都为中等程度的杂规聚合物.值得一提的是, 配体空间位阻对催化效果有较大的影响:化合物28和29催化外消旋丙交酯聚合得到了分子量分布较窄的聚合物; 而化合物27却得到了分子量分布较宽的聚合物.
2013年, Otero等[13]将配体30与ZnEt2反应生成了化合物31 (Eq. 18), 并将其用来催化外消旋丙交酯的聚合反应.发现, 31是首例可获得等规聚合物的手性锌催化剂, 且Pm值为0.77.进一步研究发现, 该聚合反应是由乙基引发的.
同年, Ma等[14]将手性配体32~36与Zn[N(SiMe3)2]2反应, 得到具有不同比例的非对映异构体混合物和一例对映体纯的锌化合物37~41 (Eq. 19), 这可能是由于配体空间位阻逐渐增大造成的.随后, 他们将这些化合物用来催化外消旋丙交酯的聚合反应, 发现, 这些催化剂都具有较高的催化活性.有趣的是, 随着催化剂中非对映异构体a的比例逐渐增加, 所得聚合物的立体选择性发生了明显的转变, 依次由杂规倾向(Pm=0.39) 转变为较高的全同选择性(Pm=0.84).此外, 通过动力学研究发现, 催化剂41能够优先聚合右旋丙交酯, 聚合过程受到了对映体的位点控制.
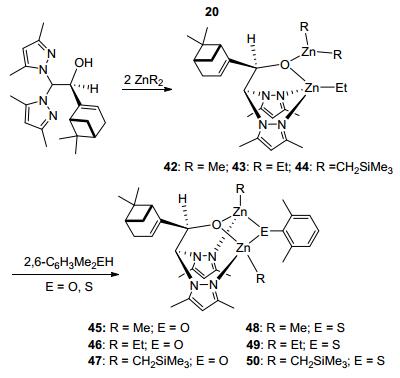
2014年, Otero等[15]将手性配体20分别与2 equiv.的烷基锌反应生成了相应的双核锌化合物42~44, 并进一步将这些烷基锌化合物与取代苯酚或取代硫酚反应, 制得了相应的衍生物45~50.随后, 他们将化合物44~47、50分别用来催化左旋丙交酯的聚合反应(Scheme 1).发现, 这些化合物都能够有效地催化丙交酯的聚合, 所得聚合物的分子量与转化率成线性关系且分子量分布较窄. MALDI-TOF质谱分析和核磁氢谱研究发现, 化合物45~47的链引发基团为酚氧基而非烷基.动力学研究表明, 聚合反应速率与单体和催化剂均成一级反应.另外, 当这些催化剂在催化外消旋丙交酯聚合反应时, 均得到了对映体点控制的等规聚合物, 且规整度随着催化剂空间位阻的增大而增大(Pm=0.59~ 0.74), 同时聚合物的熔点也随着规整度的增加而逐渐增加(Tm=150~166 ℃).
图 图式1
配合物42~50的制备
Figure 图式1.
Preparation of complexes 42~50
同年, Du等[16]将具有不同空间位阻和不同电子效应的手性胺基-噁唑啉配体分别与Zn[N(SiMe3)2]2反应, 得到相应的手性有机锌化合物51~57 (Eq. 20).之后, 他们将这些手性锌催化剂用来催化外消旋丙交酯聚合反应.发现, 它们具有较高的催化活性, 所得聚合物分子量分布较窄, 且为具有较高规整度的等规聚合物.其中, 具有吸电子取代基的催化剂57的立体选择性最高, Pm值高达0.91 (Tm=212 ℃), 这是目前为止全同选择性最高的手性锌催化剂. MALDI-TOF研究表明, N(SiMe3)2为链引发基团.
2015年, Yao等[17]将手性Schiff碱配体58~60与ZnMe2反应, 得到手性双核锌化合物61~63 (Eq. 21).随后, 他们发现这些双核锌化合物在BnOH存在下可催化外消旋丙交酯的聚合反应, 且都具有较高的催化活性, 所得聚合物的分子量分布较窄, 但是它们都未表现出明显的立体选择性, 导致无规聚合物的生成.
2 手性有机铝化合物催化的丙交酯聚合反应
手性有机铝化合物在催化丙交酯聚合反应中表现出较高的催化活性和立体选择性, 因此也获得了广泛的研究.
1996年, Spassky等[18]利用联萘胺衍生的手性配体64合成了手性甲氧基铝化合物65 (Eq. 22), 随后他们将其用于催化外消旋丙交酯的聚合反应.发现, 当转化率为19%时, 消耗掉的单体中有88%为右旋丙交酯, 表明该催化剂能够优先聚合右旋丙交酯, 显示了较高的立体选择性.当单体基本反应完全时, 得到了全同嵌段聚合物.进一步研究发现, 该反应中甲氧基为活性引发基团, 所得聚合物的分子量分布较窄.
1999年, Coates等[19]将手性配体64与Al(OiPr)3反应得到的化合物66 (Eq. 23) 用来催化内消旋丙交酯的聚合反应, 获得了具有较高规整度的间同聚合物(Pr=0.96).进一步研究发现, 该反应为单点活性可控聚合反应, 分子量分布很窄, 链增长机理为对映体的点控制机理.为了进一步研究该聚合反应机理, 2002年, 他们[3]将66与等物质的量的内消旋丙交酯进行反应, 随后将所得产物水解, 并加入(S)-α-甲氧基-α-三氟甲基-苯基乙酰氯[简写为: (S)-MTPA(Cl)]制得相应的非对映异构体Mosher酯67和68 (Scheme 2).通过氢谱分析发现, 67与68的比例为3:97, 这个比例与实验所得聚合物的立体选择性(4:96) 基本一致, 也就是说66优先从B点对内消旋丙交酯进行开环, 从而得到了高间规聚丙交酯.
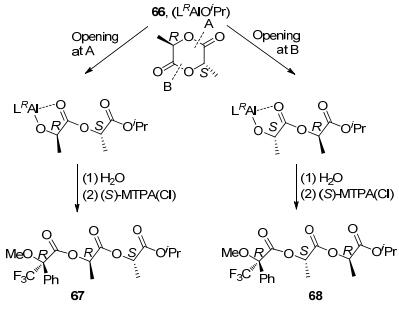 图 图式2
Mosher酯67和68的制备
Figure 图式2.
Preparation of mosher ester 67 and 68
图 图式2
Mosher酯67和68的制备
Figure 图式2.
Preparation of mosher ester 67 and 68
2002年, Feijen等[20]将手性环己二胺衍生的配体69与Al(OiPr)3反应所生成的化合物70 (Eq. 24) 用来催化外消旋丙交酯的聚合反应.发现, 不论是在溶液中还是在熔融状态下, 该催化剂都能得到规整度较高的等规聚合物(Pm>0.9), 且聚合过程为活性可控聚合.为了进一步研究该催化过程, 2003年, 他们[21]通过动力学研究发现, 催化剂70优先催化左旋丙交酯的聚合反应(kL/kD≈14), 属于对映体点控制链增长机理.
2008年, Carpentier等[22]将手性环己二胺衍生的Schiff碱配体71与Al(OiPr)3反应, 生成了相应的手性异丙氧基铝化合物72 (Eq. 25), 并将该化合物用来催化外消旋丙交酯的聚合反应.发现, 尽管该化合物的催化活性不高, 但是不论在70 ℃的甲苯溶液中, 还是在无溶剂的本体聚合中, 化合物72都能催化外消旋丙交酯的聚合反应, 且获得分子量分布相对较窄的等规嵌段聚合物(Pm>0.79).氢谱分析发现, 该聚合反应的活性引发基团为异丙氧基.
2009年, Feijen等[23]将环己二胺衍生的手性配体73~75分别与AlEt3反应, 生成了相应的手性铝化合物76~78 (Eq. 26).随后, 他们将这些化合物与等当量的异丙醇原位反应后直接用来催化丙交酯的聚合反应.发现, 聚合物的分子量分布很窄.在催化外消旋丙交酯的聚合反应中, 催化剂76~78表现出了不同的立体选择性:催化剂76可得等规聚合物(Pm=0.66), 77获得杂规聚合物(Pr=0.64), 78生成近似无规聚合物(Pr=0.55).动力学研究发现, 催化剂76能够优先聚合左旋丙交酯(kL/kD=10.1), 因此它催化外消旋丙交酯的聚合反应时可得到由对映体点控制的等规聚合物.这些催化剂76~78在催化内消旋丙交酯的聚合反应时, 均得到了具有不同规整度的间规聚合物, 说明该聚合反应是通过对映体的点控制机理来控制链增长的.
2013年, Otero等[24]将手性配体79分别与AlMe3和AlEt3反应生成了相应的化合物80和81 (Eq. 27), 并进一步将这两个化合物用来催化外消旋丙交酯的聚合反应. 110 ℃下, 甲苯中, 80和81可催化外消旋丙交酯聚合, 所得聚合物的分子量分布较窄, 但立体选择性较差, 都为无规聚合物. MALDI-TOF分析发现, 由于反应温度过高, 聚合过程中酯交换反应较为严重, 从而造成聚合物的规整度降低.另外, 研究发现, 化合物中甲基或乙基为活性引发基团.此外, 他们还将化合物80与2, 6-二甲基苯酚进行原位反应后用来催化外消旋丙交酯的聚合反应.发现, 该体系的聚合反应速率比对应的甲基化合物的快, 且聚合物的分子量分布也有所变窄.同时, 该体系在130 ℃下的本体聚合反应中也显示了较好的催化性能.但是无论是在溶液中, 还是在高温下的本体聚合反应中, 获得的仍然是无规聚合物.同样, 酚氧基为活性链引发基团.
同年, Pang等[25]将手性配体82~87与AlMe3反应生成相应的手性铝化合物88~93 (Eq. 28).随后, 在异丙醇存在下, 他们将这些化合物用来催化丙交酯的聚合反应.发现, 70 ℃下, 甲苯中, 化合物88~93可有效地催化丙交酯的聚合反应, 且所得聚合物的分子量分布很窄, 异丙氧基为活性链引发基团.另外, 他们发现配体取代基位阻越大, 则催化剂的活性越低; 配体取代基拉电子能力越强, 则催化剂的活性越高, 这是由于当配体取代基拉电子能力越强, Al-OiPr键就越弱, 从而加快了反应速率.所得聚合物均为等规聚合物, 且规整度随着配体取代基的增大而升高.反应温度对聚合物的立体选择性也有一定影响, 随着反应温度降低, 聚合物的规整度增加.动力学研究发现, 化合物93催化左旋丙交酯的聚合反应过程中, 对于单体和催化剂均为一级反应.
2014年, 我们[26]将联萘酚胺和联苯酚胺衍生的一系列手性配体分别与AlMe3或AlEt2Cl反应, 生成了相应的手性铝化合物94~103 (Eq. 29).随后在异丙醇或环氧丙烷存在下, 我们将这些化合物用来催化外消旋丙交酯的聚合反应.发现, 70 ℃下, 甲苯中, 这些化合物都可有效地催化外消旋丙交酯的聚合反应, 且获得分子量分布较窄的中等杂规聚合物.另外, 化合物94~103在甲苯中的催化反应速度均比在四氢呋喃(THF)中的快, 这是由于THF的配位效应造成的.
随后, 我们[27]又将一系列手性水杨醛Schiff碱配体分别与AlMe3或AlEt2Cl反应, 生成了相应的手性铝化合物104~114 (Eq. 30), 并进一步考查了这些化合物在外消旋丙交酯聚合反应中的催化效果.发现, 异丙醇或环氧丙烷存在下, 70 ℃, 甲苯中, 这些化合物都可有效地催化外消旋丙交酯的聚合反应, 且获得分子量分布较窄的中等杂规聚合物.同时, 还观察到, 单核铝化合物比双核铝化合物具有较高的催化活性, 这是由于空间位阻造成的.另外, 这些化合物在甲苯中的催化速度均比在THF中的快, 这是由于THF的配位效应引起的.
与此同时, Kol等[28]将手性配体115~118分别与AlMe3或Al(OiPr)3反应, 生成了相应的手性铝化合物119~124 (Scheme 3).随后, 他们将这些化合物用来催化外消旋丙交酯的聚合反应.发现, 所得聚合物的数均分子量与理论值相近, 且分子量分布都比较窄.化合物119与异丙醇原位反应后的催化效果与化合物120的催化效果相当.另外, 他们发现无论是原位反应所得催化剂还是分离所得化合物120催化外消旋丙交酯所得聚合物均为杂规聚合物, 而化合物121~124却生成了等规聚合物.说明聚合物的立构规整性与配体取代基有关.为了进一步研究该体系的反应机理, 他们以化合物122为例, 动力学研究发现, 聚合速度与单体成一级反应, 且左旋丙交酯能够优先发生聚合(kL/kD=10).该聚合过程既不是链端单一控制也不是对映体点单一控制, 而是由二者协同控制的结果, 所得聚合物为梯度全同多嵌段聚丙交酯.
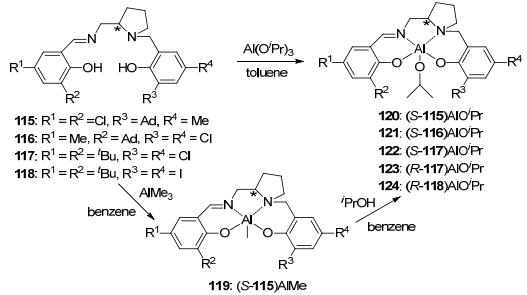 图 图式3
配合物119~124的制备
Figure 图式3.
Preparation of complexes 119~124
图 图式3
配合物119~124的制备
Figure 图式3.
Preparation of complexes 119~124
同年, Kim等[29]将手性配体125与AlMe3反应, 生成了中心金属离子具有四角锥配位构型的手性双核铝化合物126 (Eq. 31).室温下, 该化合物在异丙醇中能够稳定存在.随后他们将该化合物用来催化左旋丙交酯的聚合反应, 发现, 130 ℃下, 异丙醇存在下, 所得聚合物的数均分子量随着单体与催化剂之比的增加而线性增长, 且分子量分布都比较窄.但是, 这些聚合物的规整度都不高, 均为无规或等规倾向的聚合物, 说明单体在聚合过程中发生了异构化.
同年, Du等[30]用手性双齿配体127~135与AlMe3反应也合成了一系列手性铝化合物136~144 (Eq. 32).随后, 他们将这些化合物用来催化外消旋丙交酯的聚合反应.发现, 除化合物137、139和141外, 其它化合物在苄醇存在下都可催化丙交酯的聚合反应, 这是由于空间位阻造成的.所得聚合物的分子量分布较宽, 这是由于酯交换反应引起的. MALDI-TOF研究表明, 苄氧基为链引发基团.进一步研究发现, 随着配体上特定位置取代基空间位阻大小和电子效应的不同, 所得聚合物的规整度不同:当R1为苯基或当R3为吸电子基时, 所得聚合物为等规聚合物; 其余则为杂规聚合物.
2015年, Duan等[31]将手性Schiff碱配体145~147与Al(OiPr)3反应生成了相应的手性铝化合物148~150 (Eq. 33).随后, 他们将这些化合物用来催化丙交酯的聚合反应.发现, 化合物148~150可有效催化丙交酯的聚合反应, 且所得聚合物的分子量分布较窄, 异丙氧基为链引发基团.另外, 配体取代基位阻越大, 则催化剂的活性越低; 具有拉电子取代基的催化剂149活性最高.所得聚合物均为等规聚合物, 且规整度随着配体取代基的增大而升高.反应温度对聚合物的立体选择性也有一定影响, 随着反应温度降低, 聚合物的规整度增加.动力学研究发现, 化合物150催化左旋丙交酯的聚合反应过程中, 对于单体和催化剂均为一级反应.
3 手性稀土金属有机化合物催化的丙交酯聚合反应
稀土金属离子因其具有较强的路易斯酸性, 使得手性稀土金属有机化合物在催化丙交酯的聚合反应方面也引起了化学家们的研究兴趣.
1999年和2002年, Coates等[3, 19]将手性联萘胺Schiff碱配体64与Y(OCH2CH2NMe2)3反应生成了手性双金属钇化合物151 (Eq. 34). 70 ℃下, 该化合物在甲苯中可催化100 equiv.的内消旋丙交酯的聚合反应, 且在14 h内反应完全.但研究发现, 所得聚合物为无规聚合物, 单体的聚合过程没有选择性.
同年, Hillmyer等[32]将手性配体152~153与Y[N(SiMe3)2]3反应后所生成的化合物154和155用来催化外消旋丙交酯的聚合反应(Eq. 35).发现, 化合物155催化的聚合反应速度以及所得聚合物的数均分子量明显高于化合物154的, 说明配体结构的改变对反应活性有很大的影响.研究还发现, 化合物154和155对左旋和右旋丙交酯都没有立体选择性, 且化合物155催化左旋丙交酯的聚合反应可获得单一全同聚合物, 没有发生异构化.
2006年, Carpentier等[33]将手性噁唑啉配体156和157分别与Y[N(SiHMe2)2]3(THF)2反应生成化合物158和159 (Eq. 36), 并将这两个化合物用来催化外消旋丙交酯的聚合反应.发现, 室温下无论是在甲苯中还是THF溶液中, 化合物158和159作为单点催化剂都能够在5~10 min内快速地将100 equiv.的单体完全聚合, 表现出了较高的催化活性.聚合物的分子量分布较窄, 但是立体选择性较差, 均为无规聚合物.次年, 他们[34]将配体160分别与Y[N(SiMe3)2]3和La[N(SiHMe2)2]3-(THF)2反应生成化合物161和162 (Eq. 37), 并将这两个化合物用来催化外消旋丙交酯的聚合反应.发现, 化合物161的催化活性受溶剂的影响较大, 在甲苯中的反应活性明显比在THF中的高, 这是由于THF的配位效应引起的.另外还发现, 化合物161的催化活性远高于化合物162.所得化合物均为中等程度的等规聚合物, 且分子量分布相对较窄, 但所得数均分子量明显高于理论值, 这可能是由于催化剂的引发效率较低导致的.此外, 反应温度和溶剂对立体选择性也有一定的影响, 降低温度, 甲苯作为溶剂有利于提高聚合物的规整度.
2008年, Williams等[35]将配体163和164分别与Y[N(SiHMe2)2]3(THF)2反应所生成的化合物165和166用来催化外消旋丙交酯的聚合反应(Eq. 38).发现, 室温下化合物165和166都能够在CH2Cl2中将200 equiv.的单体在10 min之内转化完全, 表现出了较高的催化活性.但测得聚合物的分子量约为理论值的二倍, 这是由于只有一部分的N(SiHMe2)2在反应中引发了聚合反应.动力学研究发现, 化合物165和166催化外消旋丙交酯的聚合反应都经过了一定的引发期, 这是由于空间位阻较大的胺基基团延缓了引发期, 同时聚合物的分子量分布较宽, 这可能是由于较长的引发期和酯交换反应引起的. MALDI-TOF分析发现, 链增长是通过配位插入机理进行的, N(SiHMe2)2为链的活性引发基团.氢谱分析表明, 所有聚合物均为中等程度的杂规聚合物.
2008年, Zi等[36]将手性配体167分别与Y[N(SiMe3)2]3和Yb[N(SiMe3)2]3反应后所生成的化合物168和169用来催化外消旋丙交酯的聚合反应(Eq. 39).发现, 室温下, 这两个化合物都能在甲苯和THF中催化500 equiv.的单体进行聚合反应, 且在THF体系中的反应速度稍快.另外, 在THF中, 化合物168的反应活性相对较高.相反, 在甲苯中, 化合物169的反应活性相对较高.所得聚合物均为中等程度的等规聚合物, 且分子量分布较窄.
2009年, 他们[37]将联萘酚胺衍生的手性配体分别与Sm[N(SiMe3)2]3和Y[N(SiMe3)2]3反应, 生成了相应的手性化合物170和171 (Eqs. 40~41), 并进一步将其用来催化外消旋丙交酯的聚合反应.发现, 室温下, 化合物170和171在甲苯中能够催化1000 equiv.的单体且在1 h之内转化完全.相对而言, 在THF中, 这两个化合物的催化活性较低, 这可能是由于THF的配位作用引起的.另外, 化合物170在甲苯中的催化活性略低于化合物171.所得聚合物均为中等程度的等规聚合物, 且分子量分布较窄.
同年, Otero等[38]将手性配体79与Nd[N(SiH-Me2)2]3(THF)2反应, 获得了首例手性钕胺基化合物172 (Eq. 42).该化合物可催化外消旋丙交酯的聚合反应.所得聚合物的分子量分布很窄, 链引发基团为N(SiH-Me2)2.动力学研究发现, 该聚合反应速度与单体为一级反应.氢谱分析发现, 低转化率下(10%)得到的聚合物具有明显的等规性质(Pm=0.61), 且聚合物的旋光值为负值; 当转化率达到52%时, 聚合物的等规度明显下降(Pm=0.29).由此可以看出, 在较低转化率过程中, 化合物172优先聚合左旋丙交酯, 链增长为对映体的点控制机理.
2011年, Zi等[39]将手性酰胺配体173和174分别与Y[N(SiMe3)2]3反应, 得到相应的化合物175和176 (Eqs. 43, 44).室温下, 这两个化合物在甲苯中均能使1000当量的外消旋丙交酯在2 h内聚合完全, 表现出了较好的催化活性.然而, 在THF中, 两者的催化活性均有所降低, 这是由于THF的配位效应引起的.所得聚合物均为中等等规聚合物, 且分子量分布较窄.
2013年, Roesky等[40]将手性钾盐177与稀土硼氢化合物反应生成了相应的手性稀土化合物178~181 (Eq. 45), 并进一步将这些化合物用来催化外消旋丙交酯的聚合反应.发现, 60 ℃下, 化合物179~181都可催化该聚合反应, 但化合物178在此温度下没有催化活性.当温度升至80 ℃时, 化合物178才表现出了一定的催化活性, 这可能是由于钪离子半径较小造成的.同时, 这也是首例钪的硼氢化合物催化环内酯的聚合反应.化合物179~181都可催化外消旋丙交酯的聚合反应, 且聚合物的分子量分布相对较窄.但催化剂对左旋和右旋两种单体没有选择性, 所得聚合物均为无规聚合物.氢谱分析发现, 所得聚合物为α, ω-二羟基爪状聚合物, 这是由于BH -在反应中不仅是链引发基团而且还是还原剂.
2014年, 我们[41]将联萘酚胺和联苯酚胺衍生的一系列手性Schiff碱配体分别与Ln[N(SiMe3)2]3反应生成相应的化合物182~190 (Eq. 46), 并进一步将它们用来催化外消旋丙交酯的聚合反应.发现, 室温下, 单核化合物185~188在甲苯中均可使500 equiv.的丙交酯在4 h内聚合完全, 而双核化合物182~184以及189~190的催化活性相对较低, 这是由于空间位阻较大造成的.另外, 在THF中, 有机钇化合物183、186和188的催化活性明显降低, 这是由于THF的配位效应引起的.但有机钐化合物和有机镱化合物的溶剂效应不明显, 这可能是稀土金属离子与镧系金属离子之间的性质差异引起的.所获得的聚合物均为中等等规聚合物, 且分子量分布较窄.
4 手性IVB族金属有机化合物催化的丙交酯聚合反应
近年来, IVB族手性金属有机化合物也被成功地用来催化丙交酯的聚合反应.
2006年, Gibson等[42]将手性Schiff碱配体69与Ti(OiPr)4反应, 生成了钛化合物191 (Eq. 47). 70 ℃下, 该化合物在甲苯中可催化外消旋丙交酯的聚合反应, 24 h后转化率可达到57%.所得聚合物的分子量分布较窄(PDI=1.11), 且数均分子量与单体转化率成线性关系, 但立体选择性较差, 几乎为无规聚合物(Pr=0.54).动力学研究发现, 该反应经历了大约8 h的引发期, 这可能是由于第一个单体插入到Ti—iOPr键之间后形成了一个特别稳定的中间产物, 从而导致了较长的引发期.
2007年, Kim等[43]将手性Schiff碱配体192和193分别与TiCl4反应生成了相应的手性钛化合物194~196 (Scheme 4), 并将其用来催化左旋丙交酯的聚合反应.发现, 这三个钛化合物虽然缺少常见的引发基团如烷氧基、胺基等, 但它们都能够有效地催化左旋丙交酯的聚合反应, 聚合物的分子量随着单体与催化剂比例的增大而线性升高, 且具有非常窄的分子量分布.氢谱和MALDI-TOF质谱分析发现, 该催化体系中的链引发基团为Cl, 同时也发现链增长过程中伴有酯的交换反应. GPC测定结果显示, 聚合物数均分子量为理论值的一OH半, 这可能是由于化合物194~196为双点催化剂造成的.此外, 该催化体系在催化外消旋丙交酯的聚合反应时均获得了无规聚合物, 说明它们对左旋丙交酯和右旋丙交酯并未表示出明显的选择性.
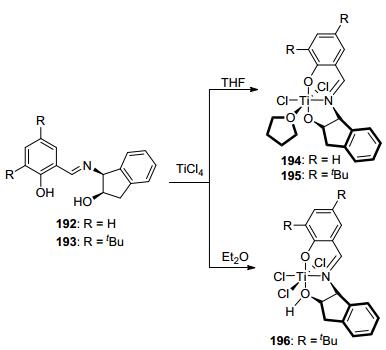 图 图式4
配合物194~196的制备
Figure 图式4.
Preparation of complexes 194~196
图 图式4
配合物194~196的制备
Figure 图式4.
Preparation of complexes 194~196
2008年, Davidson等[44]将手性Schiff碱配体197~ 200分别与Ti(OiPr)4、Zr(OiPr)4进行反应获得了相应的手性化合物201~207 (Eq. 48), 并将这些化合物用来催化外消旋丙交酯的聚合反应.发现, 在甲苯中, 手性钛化合物201和204没有催化活性, 而所有的手性锆化合物在甲苯中都表现出了较好的催化活性, 且聚合物的分子量分布较窄.另外还发现, 这些化合物不论是在固态还是在溶液中, 都产生了Λ和Δ两种非对映异构体, 因此配体所产生的手性诱导作用不大, 聚合过程中均得到了中等的杂规聚合物.改变配体的位阻大小和手性, 对反应活性和立体选择性没有明显的影响.氢谱和MALDI-TOF质谱分析表明, 异丙氧基为聚合反应的链引发基团.同时, 在无溶剂的本体聚合中, 手性钛化合物201和204都具有催化活性, 所得聚合物分子量分布较窄, 但没有选择性, 均为无规聚合物; 手性锆化合物在相同条件下得到的聚合物的分子量分布比在溶液中的宽, 但立体选择性保持不变.值得一提的是, 这些手性锆化合物尤为稳定, 不论是加入少量水进行溶液聚合, 还是直接用未经升华的单体进行本体聚合, 这些化合物都仍然具有催化活性.
2009年, Zi等[45]将手性配体208~211分别与Ti(NMe2)4反应生成了化合物212~215 (Eqs. 49~52), 并进一步将这些手性化合物用来催化外消旋丙交酯的聚合反应.发现, 化合物213~215的催化活性相似, 70 ℃下, 在甲苯或THF中可将250 equiv.的单体在24 h内转化完全, 而化合物212的催化活性较差, 这是由于空间位阻造成的.所获得的聚合物均为等规聚合物, 且分子量分布较窄.
2011年, Chakraborty等[46]将手性Schiff碱配体69分别与Zr(OiPr)4和Hf(OtBu)4反应生成了相应的手性化合物216和217 (Scheme 5).随后, 他们将手性铪化合物217与等物质的量的水反应, 生成了双金属化合物218.这三个化合物都能够高效地引发外消旋丙交酯的本体聚合反应. 140 ℃下, 化合物216~218能够在1 h内将200 equiv.的外消旋丙交酯完全转化, 所得聚合物的分子量分布很窄, 数均分子量与单体和催化剂的比例成线性关系.然而这三个催化剂对单体的立体选择性较差, 导致无规聚合物的生成, 这可能是由于化合物216~ 218的双核结构导致了链增长同时发生在在两个金属中心, 从而降低了立体选择性.通过氢谱和MALDI-TOF质谱分析发现, 烷氧基为链引发基团, 且聚合过程中酯交换反应较为普遍.动力学研究表明, 聚合速率与单体成一级反应, 且聚合反应均为瞬间引发.
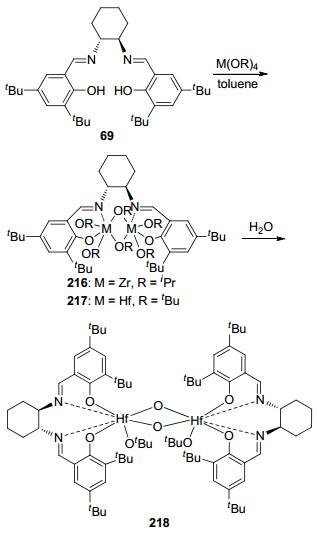 图 图式5
配合物216~218的制备
Figure 图式5.
Preparation of complexes 216~218
图 图式5
配合物216~218的制备
Figure 图式5.
Preparation of complexes 216~218
同年, Peruch等[47]将手性配体219与Ti(OiPr)4反应生成的化合物220用来催化左旋丙交酯和外消旋丙交酯的聚合反应(Eq. 53).发现, 不论是溶液聚合还是本体聚合, 化合物220都表现出了较高的催化活性.聚合物的分子量与理论值接近, 且分布较窄.通过氢谱和MALDI-TOF质谱分析发现, 聚合反应是通过配位插入机理进行的, 异丙氧基为链引发基团, 且聚合过程中有酯交换反应发生.不论在溶液中还是在无溶剂状态下的本体聚合中, 该化合物催化外消旋丙交酯的聚合反应时, 均获得无规聚合物.动力学研究发现, 左旋丙交酯和外消旋丙交酯聚合速率均与单体成一级反应, 但是二者的聚合过程都需60~70 min的引发期, 这可能是由于第一分子单体插入反应速度较慢造成的.
2014年, 我们[48]将手性咪唑盐配体221~224分别与M(NR2)4 (M=Ti, Zr, Hf; R=Me, Et)反应, 生成了相应的手性氮杂卡宾IVB族金属有机化合物225~237 (Eqs. 54~56).发现, 在异丙醇存在下, 70 ℃, 甲苯中, 这些化合物都可有效的催化外消旋丙交酯的聚合反应, 所得聚合物分子量与理论值接近, 且分子量分布较窄.动力学研究发现, 聚合反应速率与单体成一级反应, 同时聚合物的数均分子量随着转化率的升高呈线性增加.进一步研究发现, 这些化合物在甲苯中聚合反应较快, 而在THF中则较慢, 这是由于THF的配位效应造成的.此外, 有机锆和有机铪化合物的催化活性高, 而有机钛化合物的较低, 这是由于金属离子半径效应造成的.另外, 我们还观察到, 锆化合物230的催化速度比锆化合物235的催化速度快, 这是由于配体空间位阻引起的.
5 其它手性金属有机化合物催化的丙交酯聚合反应
除了上述四类主要手性金属有机化合物外, 还有一些其它手性金属有机化合物也可催化丙交酯的聚合反应.
5.1 手性碱金属有机化合物催化的丙交酯聚合反应
2010年, Lin等[49]将手性配体238与碱金属胺基化合物反应生成了三个手性碱金属化合物239~241 (Eq. 57), 并进一步将它们用来催化左旋丙交酯和外消旋丙交酯的聚合反应.发现, 相同条件下, 在这三个化合物催化左旋丙交酯的活性顺序为239<240<241, 聚合物分子量分布都比较窄.通过对化合物239催化左旋丙交酯聚合行为进行系统研究发现, 得到的数均分子量与理论值几乎一致, 且分子量分布很窄.同时, 聚合物的杂规度随着反应温度的降低而升高, -30 ℃, Pr值最高达0.82, 这可能是低温下反应速度较慢, 有利于化合物进行结构调整, 从而增加了立体选择性; 同一温度下, 当溶剂由甲苯换为THF时, 聚合物规整度稍有增加, 但是反应速度大大降低, 这可能是由于THF的配位效应引起的.
5.2 手性碱土金属有机化合物催化的丙交酯聚合反应
2000年, Chisholm等[5]利用手性铊化合物1合成了相应的手性镁化合物242 (Eq. 58), 并进一步将其用来催化丙交酯的聚合反应.发现, 在催化外消旋和内消旋丙交酯混合物的共聚反应时, 该化合物优先引发内消旋丙交酯的聚合反应且获得中等间规聚合物, 表现出了一定的立体选择性.
2012年, Okuda等[50]将手性配体243与Ca[N-(SiMe3)2]2(THF)2反应生成了手性钙化合物244 (Eq. 59), 并将其用来催化丙交酯的聚合反应.发现, 该化合物可有效地催化内消旋丙交酯的聚合反应, 获得中等间规聚合物.另外, 室温下, 在THF中, 该化合物能够催化100 equiv.的左旋丙交酯且在1 h内完全转化为全同聚合物, 没有发生异构化.
2014年, Ma等[51]将手性配体36与Mg[N(SiMe3)2]2反应, 生成一对非对映异构选择性为7:1的镁化合物245 (Eq. 60).发现, 室温下, 该化合物可催化200 equiv.的外消旋丙交酯在25 min内几乎聚合完全, 表现出较高的催化活性.令人意外的是, 该镁化合物催化外消旋丙交酯聚合获得了较高的杂规聚乳酸(Pr=0.80), 这与具有相同结构的锌化合物所表现出的立体选择性截然相反(Pm=0.81).随后, 作者对这种不同立体选择性结果进行了机理探究.他们将具有相同结构的锌和镁化合物分别与乳酸酯反应, 通过X单晶衍射分析和NMR分析, 发现, 生成的锌乳酸酯化合物具有五配位, 而镁乳酸酯化合物则具有六配位, 这一不同的立体配位环境则是造成不同选择性的原因.
2015年, 我们[52]将由戊二烯基衍生的手性配体246分别与CaI2、SrI2进行反应生成了对应的手性催化剂247和248 (Scheme 6), 并将这两个催化剂用来催化外消旋丙交酯的聚合反应.发现, 这两个化合物在不加任何活化试剂的条件下均能有效地催化外消旋丙交酯的聚合反应, 获得中等杂规聚合物, 且聚合物的分子量与理论值接近, 分子量分布较窄.通过对化合物247催化外消旋丙交酯聚合行为进行系统研究发现, 聚合物分子量随着单体转化率的升高而呈直线上升.动力学研究表明, 该聚合反应速率与单体成一级反应.
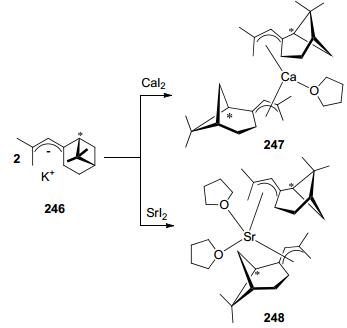 图 图式6
配合物247~248的制备
Figure 图式6.
Preparation of complexes 247~248
图 图式6
配合物247~248的制备
Figure 图式6.
Preparation of complexes 247~248
2016年, Kol等[53]将一种具有双吡咯烷骨架的手性配体249与BnMgCl反应, 生成了相应的手性镁配合物250 (Eq. 61).随后, 在BnOH存在下, 他们将其用来催化丙交酯的聚合反应.发现, 该催化剂能够快速催化次催化500 equiv.的左旋丙交酯和500 equiv.的右旋丙交酯在30 min内进行序列聚合, 获得具有高熔点(Tm=215~220 ℃)的二嵌段聚合物.此外, 它还可依次催化左旋和右旋丙交酯进行完全聚合, 生成长达六嵌段的聚乳酸.作者将该催化剂称为一种具有“量体裁衣”特性的活性聚合催化剂.
5.3 手性有机铟化合物催化的丙交酯聚合反应
2010年, Arnold等[54]将手性双齿配体251与 In[N(SiMe3)2]3并将其用来催化左旋丙交酯的开环聚合反应.发现, 该催化剂能够有效地催化左旋丙交酯的聚合反应, 且获得全同聚合物. MALDI-TOF质谱分析发现, 单体是通过配位插入机理引发的, 链的引发基团为N(SiMe3)2.
2012年, Mehrkhodavandi等[55]利用手性配体253和254合成了相应的手性铟化合物255和256 (Scheme 7), 并将这两个化合物用来催化丙交酯的聚合反应.发现, 室温下, 化合物255和256能够保持双金属结构且高效地催化外消旋丙交酯的聚合反应.将聚合物的数均分子量与理论值进行比较发现, 255为单点催化剂, 而256为双点催化剂.动力学研究表明, 这两个化合物催化的聚合过程均经过了一定时间的引发期, 链增长阶段的反应速率与单体和催化剂均成一级反应.另外还发现, 化合物255对左旋和右旋丙交酯有明显的选择性(kL/kD=14), 但是当它催化外消旋丙交酯时却得到了无规聚合物; 化合物256虽然对左旋和右旋丙交酯的选择性较低(kL/kD=2), 但在催化外消旋丙交酯时却获得了中等程度的等规聚合物.次年, 他们[56]又利用手性Schiff碱配体69合成了手性铟化合物257 (Scheme 8).该化合物可有效地催化外消旋丙交酯的聚合反应, 得到全同嵌段聚合物(Pm=0.77).将聚合物的数均分子量与理论值相比较发现, 化合物267为单点催化剂, 乙氧基为链的引发基团.动力学研究表明, 该体系的聚合速率与单体成一级反应.为了研究链的增长机理, 他们将化合物257分别用来催化左旋和右旋丙交酯的聚合反应.发现, 该催化剂能够优先聚合左旋丙交酯(kL/kD=5), 对映体的位点控制机理在链增长过程中占主导地位.
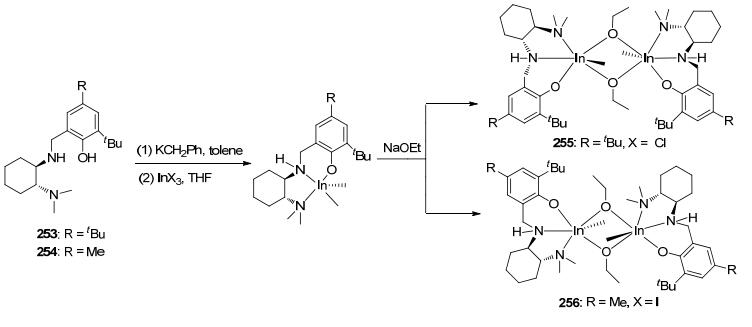 图 图式7
配合物255~256的制备
Figure 图式7.
Preparation of complexes 255~256
图 图式7
配合物255~256的制备
Figure 图式7.
Preparation of complexes 255~256
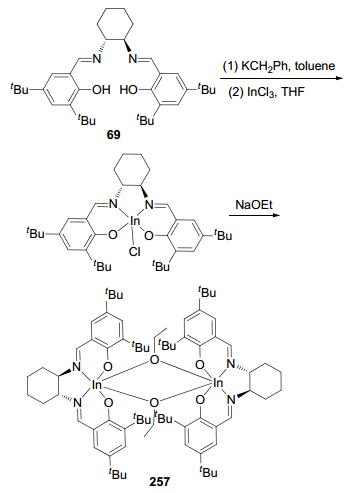 图 图式8
配合物255~256的制备
Figure 图式8.
Preparation of complexes 255~256
图 图式8
配合物255~256的制备
Figure 图式8.
Preparation of complexes 255~256
5.4 手性VB族金属有机化合物催化的丙交酯聚合反应
2013年, Pampaloni等[57]利用手性苯丙氨酸258合成了相应的手性铌、钽化合物259和260 (Eq. 63).这两个化合物都能够催化丙交酯的本体聚合反应.其中, 钽化合物260表现出了较为罕见的催化活性.相同条件下, 铌化合物259可催化单体获得较高的转化率.氢谱分析发现, 单体是通过配位插入机理引发的, OEt为链的引发基团.化合物259和260催化左旋丙交酯时获得等规聚合物, 催化外消旋丙交酯获得杂规聚合物(Pr=0.54 (259), 0.63 (260)).所得聚合物的分子量分布较宽, 在聚合过程中可能有酯的交换反应发生.
5.5 手性铁有机化合物催化的丙交酯聚合反应
2015年, Park等[58]利用手性三齿Schiff碱配体261合成出了手性铁化合物262 (Eq. 64).该化合物在催化左旋丙交酯的本体聚合反应中表现出较高活性, 且为可控聚合.但是该催化剂在催化外消旋丙交酯聚合反应中未表现出明显的立体选择性, 所得聚合物为无规聚合物. ESI-MS表明, 该催化剂在反应过程中可能生成了铁正离子化合物, 这些正离子化合物对丙交酯的聚合反应有一定的影响.
6 结束语
随着近年来人们的广泛研究, 越来越多含有不同中心金属离子的手性配合物被用来催化丙交酯的聚合反应.研究表明, 大多数手性金属配合物均能催化丙交酯的聚合反应, 但是它们所表现出来的催化活性和立体选择性差别较大, 这与中心金属离子类型以及配体空间位阻效应和电子效应都有着密切的联系.然而, 目前手性金属配合物催化外消旋丙交酯聚合反应, 生成具有高熔点全同聚丙交酯的例子相对较少, 因此通过对配体进行严格的空间调控来合成出具有高选择性的催化剂是今后研究的热点与目标.
-
-
[1]
(a) Chiellini, E.; Solaro, R. Adv. Mater. 1996, 8, 305.
(b) Uhrich, K. E.; Cannizzaro, S. M.; Langer, R. S.; Shakesheff, K. M. Chem. Rev. 1999, 99, 3181.
(c) Drumright, R. E.; Gruber, P. R.; Henton, D. E. Adv. Mater. 2000, 12, 1841.
(d) Coates, G. W. J. Chem. Soc., Dalton Trans. 2002, 467.
(e) Ghosh, S. J. Chem. Res. 2004, 241. (f) Auras, R.; Harte, B.; Selke, S. Macromol. Biosci. 2004, 4, 835.
(g) Tsuji, H. Macromol. Biosci. 2005, 5, 569.
(h) Gunatillake, P.; Mayadunne, R.; Adhikari, R. Biotechnol. Ann. Rev. 2006, 12, 301.
(i) Wu, J.; Yu, T.-L.; Chen, C.-T.; Lin, C.-C. Coord. Chem. Rev. 2006, 250, 602.
(j) Amgoune, A.; Thomas, C. M.; Carpentier, J.-F. Pure Appl. Chem. 2007, 79, 2013.
(k) Dove, A. P. Chem. Commun. 2008, 6446.
(l) Wheaton, C. A.; Hayes, P. G.; Ireland, B. J. Dalton Trans. 2009, 4832.
(m) Stanford, M. J.; Dove, A. P. Chem. Soc. Rev. 2010, 39, 486. -
[2]
(a) Zhang, G. ; Yang, J. ; Feng, X. Prog. Chem. 2000, 12, 89 (in Chinese).
(张国栋, 杨纪元, 冯新德, 化学进展, 2000, 12, 89. )
(b) Liu, F. ; Cheng, W. Polym. Bull. 2004, 6, 38 (in Chinese).
(刘凤兴, 程为庄, 高分子通报, 2004, 6, 38. )
(c) Cui, W. ; Bei, J. ; Wang, S. Polym. Bull. 2005, 5, 16 (in Chinese).
(崔文瑾, 贝建中, 王身国, 高分子通报, 2005, 5, 16. )
(d) Bai, Y. ; Huang, X. ; Lei, Z. Polym. Bull. 2006, 3, 46 (in Chinese).
(白雁斌, 黄晓琴, 雷自强, 高分子通报, 2006, 3, 46. ) -
[3]
Ovitt, T. M.; Coates, G. W. J. Am. Chem. Soc. 2002, 124, 1316. doi: 10.1021/ja012052+
-
[4]
(a) O'Keefe, B. J. ; Hillmyer, M. A. ; Tolman, W. B. J. Chem. Soc. , Dalton Trans. 2001, 2215.
(b) Dechy-Cabaret, O. ; Martin-Vaca, B. ; Bourissou, D. Chem. Rev. 2004, 104, 6147.
(c) Kamber, N. E. ; Jeong, W. ; Waymouth, R. M. ; Pratt, R. C. ; Lohmeijer, B. G. G. ; Hedrick, J. L. Chem. Rev. 2007, 107, 5813.
(d) Platel, R. H. ; Hodgson, L. M. ; Williams, C. K. Polym. Rev. 2008, 48, 11.
(e) Zhang, Z. ; Zi, G. Chin. J. Org. Chem. 2009, 29, 1059 (in Chinese).
(张站斌自国甫, 有机化学, 2009, 29, 1059. )
(f) Gu, L. ; Zhu, G. ; Song, H. ; Zi, G. Chin. J. Org. Chem. 2009, 29, 1499 (in Chinese).
(古丽娜, 祝冠彬, 宋海斌, 自国甫, 有机化学, 2009, 29, 1499. )
(g) Thomas, C. M. Chem. Soc. Rev. 2010, 39, 165.
(h) Sauer, A. ; Kapelski, A. ; Fliedel, C. ; Dagorne, S. ; Kol, M. ; Okuda, J. Dalton Trans. 2013, 42, 9007.
(i) Zhang, D. ; Zi, G. Chem. Soc. Rev. 2015, 44, 1898. -
[5]
Chisholm, M. H.; Eilerts, N. W.; Huffman, J. C.; Iyer, S. S.; Pacold, M.; Phomphrai, K. J. Am. Chem. Soc. 2000, 122, 11845. doi: 10.1021/ja002160g
-
[6]
Labourdette, G.; Lee, D. J.; Patrick, B. O.; Ezhova, M. B.; Mehrkhodavandi, P. Organometallics 2009, 28, 1309. doi: 10.1021/om800818v
-
[7]
Drouin, F.; Oguadinma, P. O.; Whitehorne, T. J. J.; Prud'homme, R. E.; Schaper, F. Organometallics 2010, 29, 2139. doi: 10.1021/om100154w
-
[8]
Börner, J.; Flörke, U.; Döring, A.; Kuckling, D.; Jones, M. D.; Steiner, M.; Breuning, M.; Herres-Pawlis, S. Inorg. Chem. Commun. 2010, 13, 369. doi: 10.1016/j.inoche.2009.12.024
-
[9]
Darensbourg, D. J.; Karroonnirun, O. Inorg. Chem. 2010, 49, 2360. doi: 10.1021/ic902271x
-
[10]
Pastor, M. F.; Whitehorne, T. J. J.; Oguadinma, P. O.; Schaper, F. Inorg. Chem. Commun. 2011, 14, 1737. doi: 10.1016/j.inoche.2011.07.018
-
[11]
Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L. F.; Tejeda, J.; Honrado, M.; Garcés, A.; Lara-Sánchez, A.; Rodríguez, A. M. Organometallics 2012, 31, 4191. doi: 10.1021/om300146n
-
[12]
(a) Zhang, Z.; Zhao, N.; Ren, W.; Chen, L.; Song, H.; Zi G. Inorg. Chem. Commun. 2012, 20, 234.
(b) Zhao, N.; Chen, L.; Ren, W.; Song, H.; Zi, G. J. Organomet. Chem. 2012, 712, 29. -
[13]
Honrado, M.; Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L. F.; Lara-Sánchez, A.; Tejeda, J.; Carrión, M. P.; Martínez-Ferrer, J.; Garcés, A.; Rodríguez, A. M. Organometallics 2013, 32, 3437. doi: 10.1021/om4003279
-
[14]
Wang, H.; Ma, H. Chem. Commun. 2013, 49, 8686. doi: 10.1039/c3cc44980g
-
[15]
Honrado, M.; Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L. F.; Garcés, A.; Lara-Sańchez, A.; Rodríguez, A. M. Organometallics 2014, 33, 1859. doi: 10.1021/om500207x
-
[16]
Abbina, S.; Du, G. ACS Macro Lett. 2014, 3, 689. doi: 10.1021/mz5002959
-
[17]
Gao, A.; Yao, W.; Xiao, Y.; Zhang, M.; Zhu, G.; Zhang, N.; Wang, S.; Wang, D.; Zhang, Y.; Gao, Y.; Xu, Z.; Lu, P.; Zhang, Z. Polyhedron 2015, 85, 537. doi: 10.1016/j.poly.2014.08.067
-
[18]
Spassky, N.; Wisniewski, M.; Pluta, C.; Borgne, A. L. Macromol. Chem. Phys. 1996, 197, 2627. doi: 10.1002/macp.1996.021970902
-
[19]
Ovitt, T. M.; Coates, G. W. J. Am. Chem. Soc. 1999, 121, 4072. doi: 10.1021/ja990088k
-
[20]
Zhong, Z.; Dijkstra, P. J.; Feijen, J. Angew. Chem., Int. Ed. 2002, 41, 4510. doi: 10.1002/1521-3773(20021202)41:23<4510::AID-ANIE4510>3.0.CO;2-L
-
[21]
Zhong, Z.; Dijkstra, P. J.; Feijen, J. J. Am. Chem. Soc. 2003, 125, 11291. doi: 10.1021/ja0347585
-
[22]
Bouyahyi, M.; Grunova, E.; Marquet, N.; Kirillov, E.; Thomas, C. M.; Roisnel, T.; Carpentier, J.-F. Organometallics 2008, 27, 5815. doi: 10.1021/om800651z
-
[23]
Du, H.; Velders, A. H.; Dijkstra, P. J.; Sun, J.; Zhong, Z.; Chen, X.; Feijen, J. Chem. Eur. J. 2009, 15, 9836. doi: 10.1002/chem.v15:38
-
[24]
Castro-Osma, J. A.; Alonso-Moreno, C.; Már quez-Segovia, I.; Otero, A.; Lara-Sánchez, A.; Fernández-Baeza, J.; Rodríguez, A. M.; Sánchez-Barba, L. F.; García-Martínez, J. C. Dalton Trans. 2013, 42, 9325. doi: 10.1039/c3dt32657h
-
[25]
Gao, B.; Duan, R.; Pang, X.; Li, X.; Qu, Z.; Tang, Z.; Zhuang, X.; Chen, X. Organometallics 2013, 32, 5435. doi: 10.1021/om400714q
-
[26]
(a) Zhao, N.; Wang, Q.; Hou, G.; Song, H.; Zi, G. J. Organomet. Chem. 2014, 754, 51.
(b) Zhao, N.; Wang, Q.; Hou, G.; Song, H.; Zi, G. Inorg. Chem. Commun. 2014, 41, 6. -
[27]
(a) Zhao, N.; Wang, Q.; Hou, G.; Song, H.; Zi, G. Inorg. Chim. Acta 2014, 413, 128.
(b) Zhao, N.; Wang, Q.; Hou, G.; Song, H.; Zi, G. Inorg. Chem. Commun. 2014, 44, 86. -
[28]
Pilone, A.; Press K.; Goldberg, I.; Kol, M.; Mazzeo, M.; Lamberti, M. J. Am. Chem. Soc. 2014, 136, 2940. doi: 10.1021/ja412798x
-
[29]
Go, M. J.; Kim, S. H.; Kang, Y. Y.; Park, H.-R.; Kim, Y.; Lee, J. Inorg. Chem. Commun. 2014, 44, 139. doi: 10.1016/j.inoche.2014.03.008
-
[30]
Bian, S.; Abbina, S.; Lu, Z.; Kolodka, E.; Du, G. Organometallics 2014, 33, 2489. doi: 10.1021/om401226j
-
[31]
Gao, B.; Li, D.; Duan, Q.; Duan, R.; Pang, X. New J. Chem. 2015, 39, 4670. doi: 10.1039/C5NJ00469A
-
[32]
Chamberlain, B. M.; Sun, Y.; Hagadorn, J. R.; Hemmesch, E. W.; Young, V. G.; Jr.; Pink, M.; Hillmyer, M. A.; Tolman, W. B. Macromolecules 1999, 32, 2400. doi: 10.1021/ma990005k
-
[33]
Alaaeddine, A.; Amgoune, A.; Thomas, C. M.; Da gorne, S.; Bellemin-Laponnaz, S.; Carpentier, J.-F. Eur. J. Inorg. Chem. 2006, 3652.
-
[34]
Heck, R.; Schulz, E.; Collin, J.; Carpentier, J.-F. J. Mol. Catal. A: Chem. 2007, 268, 163. doi: 10.1016/j.molcata.2006.12.030
-
[35]
Platel, R. H.; White, A. J. P.; Williams, C. K. Inorg. Chem. 2008, 47, 6840. doi: 10.1021/ic800419t
-
[36]
Zi, G.; Wang, Q.; Xiang, L.; Song, H. Dalton Trans. 2008, 5930.
-
[37]
Wang, Q.; Xiang, L.; Song, H.; Zi, G. J. Organomet. Chem. 2009, 694, 691. doi: 10.1016/j.jorganchem.2008.11.061
-
[38]
Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Alonso-Moreno, C.; Márquez-Segovia, I.; Sánchez-Barba, L. F.; Rodríguez, A. M. Angew. Chem., Int. Ed. 2009, 48, 2176. doi: 10.1002/anie.v48:12
-
[39]
Zhang, F.; Zhang, J.; Song, H.; Zi, G. Inorg. Chem. Commun. 2011, 14, 72. doi: 10.1016/j.inoche.2010.09.034
-
[40]
Kratsch, J.; Kuzdrowska, M.; Schmid, M.; Kazeminejad, N.; Kaub, C.; Oña-Burgos, P.; Guillaume, S. M.; Roesky, P. W. Organometallics 2013, 32, 1230. doi: 10.1021/om301011v
-
[41]
Ren, W.; Chen, L.; Zhao, N.; Wang, Q.; Hou, G.; Zi, G. J. Organomet. Chem. 2014, 758, 65. doi: 10.1016/j.jorganchem.2014.02.005
-
[42]
Gregson, C. K. A.; Blackmore, I. J.; Gibson, V. C.; Long, N. J.; Marshall, E. L.; White, A. J. P. Dalton Trans. 2006, 3134.
-
[43]
Lee, J.; Kim, Y.; Do, Y. Inorg. Chem. 2007, 46, 7701. doi: 10.1021/ic700493p
-
[44]
Chmura, A. J.; Cousins, D. M.; Davidson, M. G.; Jones, M. D.; Lunn, M. D.; Mahonc, M. F. Dalton Trans. 2008, 1437.
-
[45]
Zhang, F.; Song, H.; Zi, G. J. Organomet. Chem. 2010, 695, 1993. doi: 10.1016/j.jorganchem.2010.05.003
-
[46]
Saha, T. K.; Ramkumar, V.; Chakraborty, D. Inorg. Chem. 2011, 50, 2720. doi: 10.1021/ic1025262
-
[47]
Deivasagayam, D.; Peruch, F. Polymer 2011, 52, 4686. doi: 10.1016/j.polymer.2011.07.049
-
[48]
Zhao, N.; Hou, G.; Deng, X.; Zi, G.; Walter, M. D. Dalton Trans. 2014, 43, 8261. doi: 10.1039/C4DT00510D
-
[49]
Peng, Y.-L.; Huang, Y.; Chuang, H.-J.; Kuo, C.-Y.; Lin, C.-C. Polymer 2010, 51, 4329. doi: 10.1016/j.polymer.2010.07.016
-
[50]
Davin, J. P.; Buffet, J.-C.; Spaniol, T. P.; Okuda, J. Dalton Trans. 2012, 41, 12612. doi: 10.1039/c2dt31309j
-
[51]
Wang, H.; Yang, Y.; Ma, H. Macromolecules 2014, 47, 7750. doi: 10.1021/ma501896r
-
[52]
Fecker, A. C.; Freytag, M.; Jones, P. G.; Zhao, N.; Zi, G.; Walter, M. D. Dalton Trans. 2015, 44, 16325. doi: 10.1039/C5DT02851E
-
[53]
Rosen, T.; Goldberg, I.; Venditto, V.; Kol, M. J. Am. Chem. Soc. 2016, 138, 12041. doi: 10.1021/jacs.6b07287
-
[54]
Buffet, J.-C.; Okuda, J.; Arnold, P. L. Inorg. Chem. 2010, 49, 419. doi: 10.1021/ic900740n
-
[55]
Yu, I.; Acosta-Ramírez, A.; Mehrkhodavandi, P. J. Am. Chem. Soc. 2012, 134, 12758. doi: 10.1021/ja3048046
-
[56]
Aluthge, D. C.; Patrick, B. O.; Mehrkhodavandi, P. Chem. Commun. 2013, 49, 4295. doi: 10.1039/C2CC33519K
-
[57]
Hayatifar, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S. Inorg. Chem. 2013, 52, 4017. doi: 10.1021/ic4000654
-
[58]
Kang, Y. Y.; Park, H.-R.; Lee, M. H.; An, J.; Kim, Y.; Lee, J. Polyhedron 2015, 95, 24. doi: 10.1016/j.poly.2015.04.006
-
[1]
-


 下载:
下载:







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 2706
- HTML全文浏览量: 1093
 下载:
下载:
