Received Date:
28 June 2020 Revised Date:
29 July 2020 Available Online:
01 August 2020
Fund Project:
The project was supported by the National Youth Science Foundation of China (21802158) and National Natural Science Foundation of China (21776294)
Abstract:
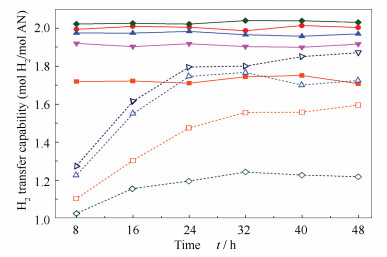
A series of boron doped reduced graphene oxide catalysts were prepared and applied to anthracene hydrogenation. The results show that, with the change of the treatment temperature of the catalyst, the ordered carbon structure in the catalyst changed and boron replaced the carbon in the material skeleton, which affected the adsorption and activation of anthracene and hydrogen. After boron doping, the catalyst showed higher activity for anthracene hydrogenation reaction, the highest conversion of anthracene was up to 97%, and the highest selectivity of deep hydrogenation product octahydroanthracene was up to 19%.
SU C L, LOH K P. Carbocatalysts:Graphene oxide and its derivatives[J]. Acc Chem Res,
2013, 46(10):
2275-2285.
doi: 10.1021/ar300118v
[2]
ZHANG Y, ZHANG L Y, ZHOU C W. Review of chemical vapor deposition of graphene and related applications[J]. Acc Chem Res,
2013, 46(10):
2329-2339.
doi: 10.1021/ar300203n
[3]
NAVALON S, DHAKSHINAMOORTHY A, ALVARO M, GARCIA H. Carbocatalysis by graphene-based materials[J]. Chem Rev,
2014, 114(12):
6179-6212.
doi: 10.1021/cr4007347
[4]
FURIMSKY E. Carbons and carbon supported catalysts in hydroprocessing[J]. Platinum Metals Rev,
2009, 53(3):
135-137.
doi: 10.1595/147106709X465479
[5]
ZHANG Z G, OKADA K, YAMAMOTO M, YOSHIDA T. Hydrogenation of anthracene over active carbon-supported nickel catalyst[J]. Catal Today,
1998, 45(1):
361-366.
[6]
SUN L B, WEI X Y, LIU X Q, ZONG Z M, LI W, KOU J H. Selective hydrogen transfer to anthracene and its derivatives over an activated carbon[J]. Energy Fuels,
2009, 23(10):
4877-4882.
doi: 10.1021/ef900398g
[7]
SUN L B, ZONG Z M, KOU J H, ZHANG L F, NI Z H, YU G Y, CHEN H, WEI X Y. Activated carbon-catalyzed hydrogenation of polycyclic arenes[J]. Energy Fuels,
2004, 18(5):
1500-1504.
doi: 10.1021/ef049946a
[8]
HUANG X, QI X Y, BOEY F, ZHANG H. Graphene-based composites[J]. Chem Soc Rev,
2012, 41(2):
666-686.
doi: 10.1039/C1CS15078B
[9]
TRANDAFIR M M, FLOREA M, NEAŢU F, PRIMO A, PARVULESCU V I, GARCÍA H. Graphene from alginate pyrolysis as a metal-free catalyst for hydrogenation of nitro compounds[J]. ChemSusChem,
2016, 9(13):
1565-1569.
doi: 10.1002/cssc.201600197
[10]
PRIMO A, NEATU F, FLOREA M, PARVULESCU V, GARCIA H. Graphenes in the absence of metals as carbocatalysts for selective acetylene hydrogenation and alkene hydrogenation[J]. Nat Commun,
2014, 5:
5291.
doi: 10.1038/ncomms6291
[11]
YANG J H, SUN G, GAO Y J, ZHAO H B, TANG P, TAN J, LU A H, MA D. Direct catalytic oxidation of benzene to phenol over metal-free graphene-based catalyst[J]. Energy Environ Sci,
2013, 6(3):
793-798.
doi: 10.1039/c3ee23623d
[12]
KONG X, SUN Z, CHEN M, CHEN C, CHEN Q. Metal-free catalytic reduction of 4-nitrophenol to 4-aminophenol by N-doped graphene[J]. Energy Environ Sci,
2013, 6(11):
3260-3266.
doi: 10.1039/c3ee40918j
[13]
刘仁厚.石墨烯基催化剂的制备及其蒽催化加氢性能研究[D].太原: 中国科学院山西煤炭化学研究所, 2017.LIU Ren-hou. Study of Graphene-based carbocatalysts for hydrogenation of anthracene[D]. Taiyuan: Institute of Coal Chemistry, Chinese Academy of Sciences, 2017.
[14]
QU L T, LIU Y, BAEK J B, DAI L M. Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells[J]. ACS Nano,
2010, 4(3):
1321-1326.
doi: 10.1021/nn901850u
[15]
WANG H B, MAIYALAGAN T, WANG X. Review on recent progress in nitrogen-doped graphene:Synthesis, characterization, and its potential applications[J]. ACS Catal,
2012, 2(5):
781-794.
doi: 10.1021/cs200652y
[16]
WEI Z Z, WANG J, MAO S J, SU D F, JIN H Y, WANG Y H, XU F, LI H R, WANG Y. In situ-generated Co0-Co3O4/N-doped carbon nanotubes hybrids as efficient and chemoselective catalysts for hydrogenation of nitroarenes[J]. ACS Catal,
2015, 5(8):
4783-4789.
doi: 10.1021/acscatal.5b00737
[17]
WU Z S, WINTER A, CHEN L, SUN Y, TURCHANIN A, FENG X L, MVLLEN K. Three-dimensional nitrogen and boron Co-doped graphene for high-performance all-solid-state supercapacitors[J]. Adv Mater,
2012, 24(37):
5130-5135.
doi: 10.1002/adma.201201948
[18]
ZHANG C H, FU L, LIU N, LIU M H, WANG Y Y, LIU Z F. Synthesis of nitrogen-doped graphene using embedded carbon and nitrogen sources[J]. Adv Mater,
2011, 23(8):
1020-1024.
doi: 10.1002/adma.201004110
[19]
LIN Z Y, WALLER G, LIU Y, LIU M, WONG C P. Facile synthesis of nitrogen-doped graphene via pyrolysis of graphene oxide and urea, and its electrocatalytic activity toward the oxygen-reduction reaction[J]. Adv Energy Mater,
2012, 2(7):
884-888.
doi: 10.1002/aenm.201200038
[20]
YAZDI A Z, CHIZARI K, JALILOV A S, TOUR J, SUNDARARAJ U. Helical and dendritic unzipping of carbon nanotubes:a route to nitrogen-doped graphene nanoribbons[J]. ACS Nano,
2015, 9(6):
5833-5845.
doi: 10.1021/acsnano.5b02197
[21]
WANG Y, SHAO Y Y, MATSON D W, LI J H, LIN X H. Nitrogen-doped graphene and its application in electrochemical biosensing[J]. ACS Nano,
2010, 4(4):
1790-1798.
doi: 10.1021/nn100315s
[22]
SHENG Z H, SHAO L, CHEN J J, BAO W J, WANG F B, XIA X H. Catalyst-free synthesis of nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis[J]. ACS Nano,
2011, 5(6):
4350-4358.
doi: 10.1021/nn103584t
[23]
SUBRAHMANYAM K S, PANCHAKARLA L S, GOVINDARAJ A, RAO C N R. Simple method of preparing graphene flakes by an arc-discharge method[J]. J Phys Chem C,
2009, 113(11):
4257-4259.
doi: 10.1021/jp900791y
[24]
ENOUZ S, STÉPHAN O, COCHON J L, COLLIEX C, LOISEAU A. C-BN patterned single-walled nanotubes synthesized by laser vaporization[J]. Nano Lett,
2007, 7(7):
1856-1862.
doi: 10.1021/nl070327z
[25]
SUENAGA K, COLLIEX C, DEMONCY N, LOISEAU A, PASCARD H, WILLAIME F. Synthesis of nanoparticles and nanotubes with well-separated layers of boron nitride and carbon[J]. Science,
1997, 278(5338):
653-655.
doi: 10.1126/science.278.5338.653
[26]
IAMPRASERTKUN P, KRITTAYAVATHANANON A, SAWANGPHRUK M. N-doped reduced graphene oxide aerogel coated on carboxyl-modified carbon fiber paper for high-performance ionic-liquid supercapacitors[J]. Carbon,
2016, 102:
455-461.
doi: 10.1016/j.carbon.2015.12.092
[27]
ZHAO Y, YANG L J, CHEN S, WANG X Z, MA Y W, WU Q, JIANG Y F, QIAN W J, HU Z. Can boron and nitrogen co-doping improve oxygen reduction reaction activity of carbon nanotubes?[J]. J Am Chem Soc,
2013, 135(4):
1201-1204.
doi: 10.1021/ja310566z
[28]
THIRUMAL V, PANDURANGAN A, JAYAVEL R, KRISHNAMOORTHI S R, ILANGOVANET R. Synthesis of nitrogen doped coiled double walled carbon nanotubes by chemical vapor deposition method for supercapacitor applications[J]. Current Appl Phys,
2016, 16(8):
816-825.
doi: 10.1016/j.cap.2016.04.018
[29]
I X H, ANTONIETTI M. Polycondensation of boron- and nitrogen-Codoped holey graphene monoliths from molecules:Carbocatalysts for selective oxidation[J]. Angew Chem Int Ed,
2013, 52(17):
4572-4576.
doi: 10.1002/anie.201209320
[30]
WANG H, ZHENG X L, CHEN H N, YAN K Y, ZHU Z L, YANG S H. The nanoscale carbon p-n junction between carbon nanotubes and N, B-codoped holey graphene enhances the catalytic activity towards selective oxidation[J]. Chem Commu,
2014, 50(56):
7517-7520.
doi: 10.1039/C4CC01707B
[31]
TANG Y B, YIN L C, YANG Y, BO X H, CAO Y L, WANG H E, ZHANG W J, BELLO I, LEE S T, CHENG H M, LEE C S. Tunable band gaps and p-type transport properties of boron-doped graphenes by controllable ion doping using reactive microwave plasma[J]. ACS Nano,
2012, 6(3):
1970-1978.
doi: 10.1021/nn3005262
[32]
PANCHAKARLA L S, SUBRAHMANYAM K S, SAHA S K, GOVINDARAJ A, KRISHNAMURTHY H R, WAGHMARE U V, RAO C N R. Synthesis, structure, and properties of boron- and nitrogen-doped graphene[J]. Adv Mater,
2009, 21:
4726-4730.
[33]
XU Z, LU W G, WANG W L, GU C Z, LIU K H, BAI X D, WANG E G, DAI H J. Converting metallic single-walled carbon nanotubes into semiconductors by boron/nitrogen Co-doping[J]. Adv Mater,
2008, 20(19):
3615-3619.
doi: 10.1002/adma.200800830
[34]
PULLAMSETTY A, SUNDARA R. Investigation of catalytic activity towards oxygen reduction reaction of Pt dispersed on boron doped graphene in acid medium[J]. J Colloid Interf Sci,
2016, 479:
260-270.
doi: 10.1016/j.jcis.2016.06.069
[35]
SHENG Z H, GAO H L, BAO W J, WANG F B, XIA X H. Synthesis of boron doped graphene for oxygen reduction reaction in fuel cells[J]. J Mater Chem,
2012, 22(2):
390-395.
doi: 10.1039/C1JM14694G
[36]
THIRUMAL V, PANDURANGAN A, JAYAVEL R, ILANGOVAN R. Synthesis and characterization of boron doped graphene nanosheets for supercapacitor applications[J]. Synthetic Met,
2016, 220:
524-532.
doi: 10.1016/j.synthmet.2016.07.011
[37]
YANG X X, LIU L, WU M H, WANG W L, BAI X D, WANG E G. Wet-chemistry-assisted nanotube-substitution reaction for high-efficiency and bulk-quantity synthesis of boron- and nitrogen-codoped single-walled carbon nanotubes[J]. J Am Chem Soc,
2011, 133(34):
13216-13219.
doi: 10.1021/ja202234z
[38]
BANHART F, KOTAKOSKI J, KRASHENINNIKOV A V. Structural defects in graphene[J]. ACS Nano,
2010, 5(1):
26-41.


 下载:
下载:
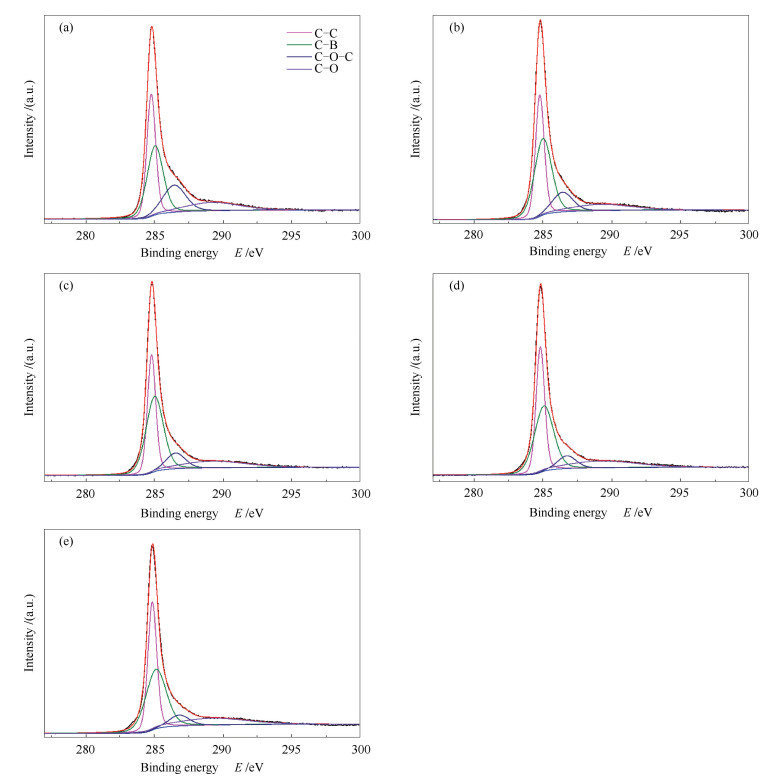
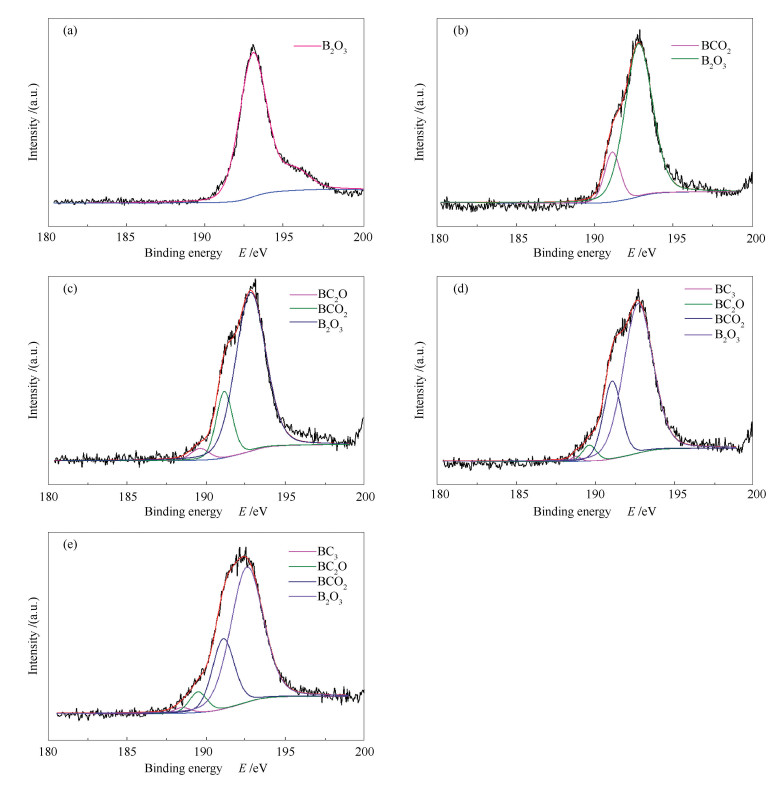
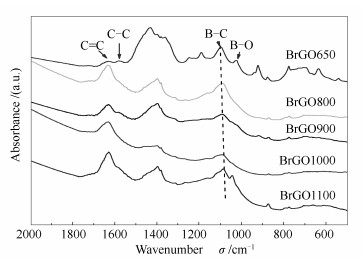
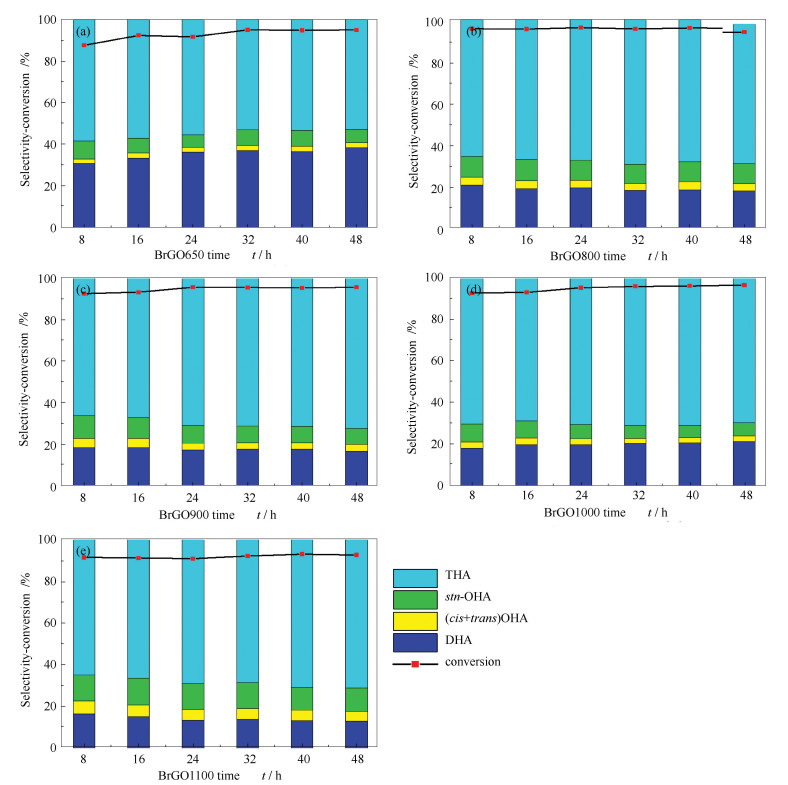

 下载:
下载:











