图 1.
单齿亚磷酸配体和二亚磷酸酯配体
Figure 1.
Monodentate phosphite ligands and diphosphite ligands

随着人们对手性化合物及手性现象认识的提高, 使得光学纯物质的合成在医药、农药、香精香料及精细化工等方面占据着重要的地位[1, 2].手性化合物主要可从天然产物中提取或通过外消旋体拆分、化学合成和催化不对称合成等方法制备得到.其中催化不对称合成具有手性增值、高对映选择性、原子经济性和易于实现工业化等特点, 它是众多不对称合成方法中最为理想的一种方法.
近几十年来, 不对称催化氢化反应、不对称催化氧化反应及过渡金属催化不对称开环反应是不对称催化反应领域的研究热点.其中, 不对称催化氢化反应被应用于工业不对称合成反应[3], 有着经济、高效和清洁等特点. 1968年, Knowles等[4]报道了手性铑-膦配合物催化剂催化的不对称氢化反应; 1974年, 美国孟都山公司将不对称氢化反应技术应用于L-多巴的工业化生产; 1980年, Noyori等[5]合成了配体2, 2'-双(二苯基膦基)- 1, 1'-联萘(BINAP), 并将该配体应用于不对称氢化反应中, 得到对映体过量值高大于99% ee的氢化产物[6].
目前, 在不对称催化氢化反应中, 含过渡金属钌[7]、铑[8]、铱[9]、铂[10]和钯[11]的催化剂比较常见.其中, 钌原子的电子结构为4d75s1, 是氧化态最多的元素, 不同的氧化态具有不同的电子结构, 一种电子结构又可能具有多种几何结构, 这为合成多种钌配合物提供良好的基础, 钌配合物还可以作为加氢催化剂[12], 因此钌配合物被广泛地应用于C=C、C=O和C=N的氢化反应[13~15]; β-酮酸酯通过不对称氢化反应得到的光学产物β-羟基酸酯可作为合成多种物质的手性原料, 具有重要的研究价值.钌、铑和铱催化体系在β-酮酸酯不对称氢化反应中都表现出较好的催化性能; 而钯催化体系在β-酮酸酯不对称氢化反应中活性较差, 所需催化剂的量较大.本文主要综述了近十年来钌催化剂分别在均相和多相两种体系中, 催化β-酮酸酯不对称氢化反应的研究进展, 并对可能的部分反应机理进行了讨论.
均相不对称氢化反应是以过渡金属与手性配体形成的配合物为催化剂, 且该催化剂可溶于反应体系, 具有高活性和高选择性等特点; 手性配体是手性催化剂产生手性诱导和立体化学控制的源泉, 并对催化剂催化活性与对映选择性有重要作用, 因此设计和合成新型高效的手性配体成为了不对称催化领域的研究热点[16].
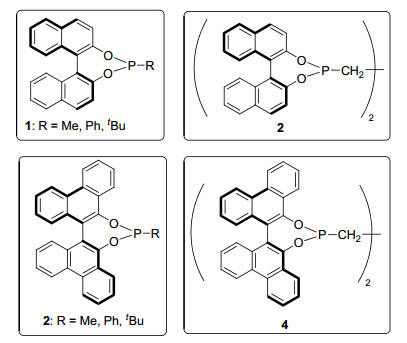
手性单膦配体是第一类被应用于不对称氢化反应的配体, 但因其对映选择性不好, 未能引起人们的关注[17]. 2000年, Pringle等[18]比较了单齿亚磷酸配体1, 2和二亚磷酸酯配体3, 4在铑催化不对称氢化反应中的对映选择性, 发现单齿体系作为催化剂时可获得更高的ee值(高达92% ee).至此单膦配体在不对称氢化反应中开始复兴(图 1).
2009年, Wasserscheid等[19]以单膦配体5与钌配位形成的配合物为催化剂, 催化乙酰乙酸甲酯的不对称氢化反应(Eq. 1).研究发现, 该反应的活化能在74.8~85.6 kJ•mol-1之间; 此外, 作者还分别研究了反应温度和氢气压力对反应的对映选择性的影响, 当升高反应温度或增加氢气压力时, 氢化产物3-羟基丁酸甲酯的ee值会升高(95% ee).
与单膦配体相比, 手性双膦配体具有配位能力强、可提高中心金属原子的碱性、降低配合物分子间或分子内的交换过程、可形成多种结构的配体和有较高的对映选择等优点, 因此在不对称催化领域占据重要地位[20].
|
|
(1) |
2008年, Börner等[21]报道了以Ru/6为催化剂, 催化3, 3-二甲氧基丙酰乙酸乙酯的不对称氢化反应(Eq. 2), 得到具有高对映选择性的3-羟基-5, 5-二甲氧基戊酸乙酯(98.7% ee).研究发现:当反应温度升高到70 ℃时, 反应速率增加10倍, 产物对映体过量值仍高于95% ee.
|
|
(2) |
2008年, Hamada课题组[22]以Ru/7为催化剂, 首次通过动态动力学拆分的方法实现α-氨基-β-酮酸酯8的不对称氢化反应(Eq. 3), 得到氢化产物9 (anti/syn=99/1).研究发现:底物8 C4位置取代基的体积对氢化产物的ee值有一定影响, 当取代基为仲烷基时, 氢化产物对映体过量值高达97% ee, 产率为86%;当取代基为叔丁基时, 氢化产物对映体过量值仅有60% ee, 产率为67%.用手性配体(R)-2, 2'-双(二苯基膦基)-6, 6'-二甲氧基-1, 1'-联苯[(R)-MeO-BIPHEP] (10)代替(S)- BINAP (7), 可提高反应的对映选择性.
|
|
(3) |
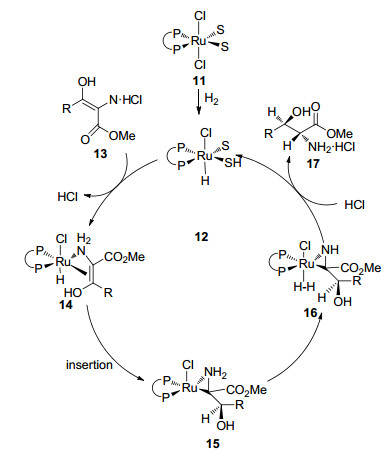
同年, 该课题组[23]通过使用氘代标记的底物进一步研究了该反应的机理(Scheme 1), 首先将RuCl2((S)- BINAP)络合物11氢化形成单氢化物配合物12, 然后12通过配体交换, 再与底物的烯醇互变异构体13反应, 生成络合物14, 然后将14烯醇-双键插入Ru—H键中得到配合物15, 15与氢气发生δ-复分解反应生成β-羟基- α-氨基酸酯17和12.络合物15经HCl质子化可以生成17和11.
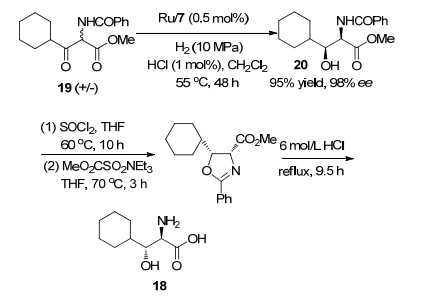
2009年, 该课题组[24]通过不对称氢化反应合成了(2R, 3R)-2-氨基-3-环己基-3-羟基丙酸(18).其中关键步骤是Ru/7催化α-氨基-β-酮酸酯(19)的不对称氢化反应(Scheme 2), 得到氢化产物20 (98% ee, 产率95%). 20经一系列反应后即可得到目标化合物18.
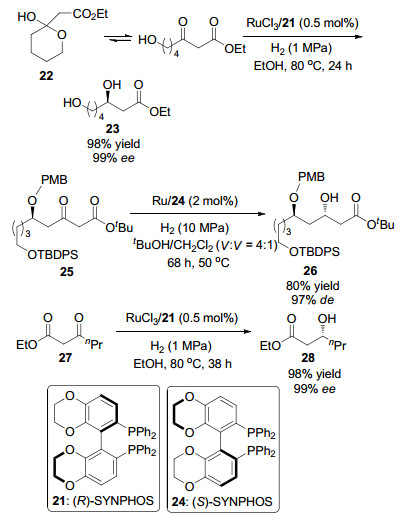
2008年, Genêt课题组[25]通过手性配体(R)-(+)-6, 6'-双(二苯基磷)-2, 2', 3, 3'-四氢-5, 5'-二-1, 4-苯并二辛[(R)-SYNPHOS] (21)与钌配位形成的催化体系, 催化β-酮酸酯不对称氢化反应, 并将该反应应用于大环内酯dolabelide A C15~C30片段的构建, 得到具有高对映选择性和非对映选择性的C19, C21和C27羟基立体中心. (1) C19羟基立体中心:以(R)-SYNPHOS (21)为手性配体, 以RuCl3为催化剂, 催化22的不对称氢化反应(Scheme 3), 得到β-羟基酯23 (99% ee); (2) C21羟基立体中心:以Ru/24为催化剂, 催化β-酮酸酯25的不对称氢化反应, 得到β-羟基酯26 (97% de); (3) C27羟基立体中心: RuCl3/21催化β-酮酸酯27生成具有高对映选择性的28 (99% ee) (Scheme 3).
2009年, Genêt等[26]将β-酮酸酯的不对称氢化反应应用于圆皮海绵内酯C3, C5和C11羟基立体中心的构建, 关键步骤为: (1)以Ru(21)Br2为催化剂, 催化β-酮酸酯29的不对称氢化反应(Eq. 4), 得到目标产物30 (99% de), 产率高达96%; (2)以[RuCl(21)(μ-Cl)3]•[Me2NH2]为催化剂, 催化β-酮酸酯31的不对称氢化反应(Eq. 5), 得到目标产物32 (99% de), 产率高达94%.
|
|
(4) |
|
|
(5) |
2010年, 该课题组[27]在配体(R)-5, 5'-双(二苯基磷)- 2, 2, 2', 2'-四氟-4, 4'-二-1, 3-苯二氧杂环[(R)-DIFLUO-RPHOS, 33]与钌形成的催化体系中, 以二氯甲烷(DCM)为溶剂, 催化2-氯-3-氧代-3-苯基丙酸甲酯的不对称氢化反应(Eq. 6), 得到(2R, 3R)-2-氯-3-羟基-3-苯基丙酸甲酯(96% de, 92% ee), 转化率只有58%;而以(R)-BINAP (6), (R)-SYNPHOS (21)或(R)-MeO-BIPHEP (10)为配体时, 反应转化率有所提高(88%, 90%, 89%), 但产物非对映体选择性明显下降(67% de, 26% de, 57% de).为提高反应转化率, 作者对溶剂进行了筛选, 该反应在甲苯或四氢呋喃(THF)中进行时, 转化率偏低(21%~25%); 在乙醚和己烷中获得了中等转化率(73%~75%)、较低的非对映选择性(10%~22% de)和中等的对映选择性(85% ee); 在1, 2-二氯乙烷(DCE)中虽然转化率较低(36%), 但de值与ee值与在二氯甲烷中得到的结果相当(94% de, 93% ee).他们得出结论:二氯甲烷为该氢化反应的最佳溶剂.
|
|
(6) |
2011年, Prévost等[28]报道了共聚酰胺的全合成.该合成依赖于两种方法: (1) α-酮基-β-炔烃酯的立体选择性还原; (2)通过钌介导的不对称氢化动力学拆分α-氨基-β-酮酸酯.作者以Ru(21)Br2或Ru(24)Br2为催化剂, 催化α-氨基- β-酮酸酯盐酸盐34的不对称氢化反应(Eq. 7), 得到目标产物氨基醇35 (96% ee, 95% de, 产率59%~66%); 在二氯甲烷溶剂中, 使用相同的催化剂, 催化α-氨基-β-酮酸酯36的不对称氢化反应(Eq. 8), 得到氢化产物37 (98% ee, 98% de, 产率82%~88%).
|
|
(7) |
|
|
(8) |
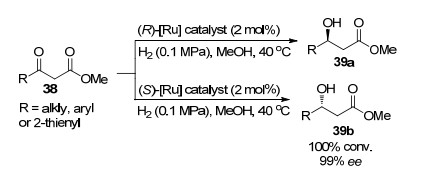
2008年, Hassine等[29]通过不对称氢化反应合成了手性1, 3-二醇和1-取代-1-丙醇, 关键步骤为:以双膦配体MeO-BIPHEP, SYNPHOS与钌形成的配合物为催化剂, 催化β-酮酸酯38的不对称氢化反应(Scheme 4), 可完全转化为具有高对映选择性的β-羟基酯39 (99% ee). β-羟基酯39经进一步反应即可得到1, 3-二醇和1-取代- 1-丙醇.
同年, Kesselgruber等[30]合成了新型芳基二膦配体(S)-(+)-N, N′-二甲基-7, 7'-双(二苯基膦)-3, 3', 4, 4'-四氢- 8, 8'-二-2H-1, 4-苯并嗪[(S)-Solphos, 40].研究发现:该类配体在钌催化β-酮酸酯41的不对称氢化反应中表现出高对映选择性(95%~99% ee) (Eq. 9), 与以SYNPHOS[29], MeO- BIPHEP[32]或Segphos[39]为配体时获得的ee值相当, 但Ru/Solphos体系的活性更高(S/C=100000).底物42的R1和R2基团与Solphos 40的Ar基团的变化对氢化产物的对映选择性影响不大.
|
|
(9) |
2008年, 陈华课题组[31]合成了新型联吡啶手性双膦配体(R)-2, 2', 6, 6'-四甲氧基-4, 4'-二(二(对甲氧基)苯基膦基)-3, 3'-联吡啶)[(R)-MeO-P-Phos, 43], 并将其应用于β-酮酸酯41的不对称加氢反应(Eq. 10).研究发现:当底物为41a时, 目标产物42a的对映体过量值为99.1% ee; 当底物取代基为吸电子基(41b)时, 羰基氧原子上的电子云密度会降低, 不利于底物分子与中心金属络合, 使两个非对映异构体之间的活化能差降低, 从而降低目标产物42b的对映选择性(57.6% ee); 且质子溶剂有利于提高反应活性和对映选择性, 乙酰乙酸甲酯在乙醇中的氢化反应, 1 h转化率即可达到100%, 氢化产物的对映体过量值高达99.1% ee; 而在甲苯溶剂中反应时, ee值仅有41.9%, 转化率为27.7%.
|
|
(10) |
2010年, 该课题组[32]以Ru(44)(C6H6)Cl2为催化剂, 催化β-酮酸酯的不对称氢化反应(Eq. 11).结果表明:在配体44中引入长链烷氧基时(44a), 对其空间效应影响较大, 可显著提高产物的对映选择性(98% ee); 当底物羰基上的取代基R1为吸电子基(41c)时, 氢化产物42c的对映选择性明显下降(74.8% ee); 由于底物41d的R1基团空间位阻较大, 与双膦配体的芳环之间存在较大的排斥作用, 不利于底物与中心金属络合, 因而反应活性较差(48.8% ee).
|
|
(11) |
2009年, 张绪穆课题组[33]合成了C(3)*-TunePhos手性双膦配体45a, 45b, 并研究了45a~45e在钌催化苯甲酰乙酸乙酯氢化反应中的对映选择性(Eq. 12).对配体45取代基的电子效应和空间效应的研究表明:引入体积较小的3, 5-二甲基(45b)和4-甲基(45c)时, 目标产物(R)-3-羟基-3-苯基丙酸乙酯的对映体过量值最高(98% ee); 引入体积较大的3, 5-二叔丁基(45d)和4-甲氧基-3, 5-二叔丁基(45e)时, 目标产物(R)-3-羟基-3-苯基丙酸乙酯的对映选择性明显降低(89% ee, 56% ee).且45b在其他β-芳基-β-酮酸酯和乙酰乙酸烷基酯的不对称氢化反应中, 均表现出较高的对映体选择性(95%~99% ee).
|
|
(17) |
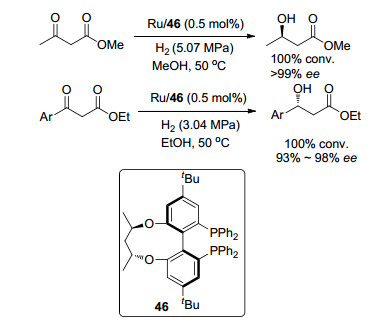
同年, 王春江和张绪穆等[34]合成了4, 4'-二叔丁基TunePhos型手性双膦配体46, 并将其应用于钌催化β-酮酸酯不对称氢化反应中.底物为乙酰乙酸甲酯时, 得到具有高对映体选择性的(R)-3-羟基丁酸甲酯(>99% ee) (Scheme 5); 该催化剂在β-芳基-β-酮酸酯的氢化反应中也表现出较高的催化活性(93%~98% ee) (Scheme 5), 与用Ru/BINAP体系获得的结果相当, 且高于Ru/C(3)-TunePhos体系[35], 说明叔丁基的引入对ee值产生了影响.
同年, 该课题组[36]报道了配体47a~47c的合成方法, 并研究了该配体在不对称氢化中的对映选择性(表 1), 作者发现这三种配体对苯甲酰乙酸乙酯的不对称氢化都是有效的, 都可生成R构型的产物, 但与C(3)*- TunePhos配体(45b, 45c)相比[33], 47a~47c的对映选择性较差.当配体为47b时, 目标产物(R)-3-羟基-3-苯基丙酸乙酯的对映选择性最高(89% ee).由于配体47a~47c都含(S)-BIPHEP部分, 他们推断, 这些配体所含的BIPHEP部分对氢化反应产物的绝对构型影响较大; 配体中的联萘部分不仅限制了BIPHEP骨架的自由旋转, 而且为催化氢化提供了独特的手性环境.
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | L* | Conv./% | ee/% | Config. |
| 1 |  |
100 | 78 | R |
| 2 |  |
100 | 83 | R |
| 3 |  |
100 | 89 | R |
2009年, 袁伟成等[37]制备了7, 7'-二取代的BINAP配体48a~48c, 这些配体在乙酰乙酸乙酯的氢化反应中表现出较高的对映选择性(Eq. 13), 氢化产物3-羟基丁酸乙酯的ee值高达98% ee.这些配体在β-酮酸酯加氢反应中, 均表现出与配体BINAP相当的对映选择性.
|
|
(13) |
2009年, Fukuzawa等[38]以49为手性配体, 以[Ru-(COD)(metallyl)2]为催化剂, 催化2-氧代环戊烷羧酸乙酯的不对称氢化反应(Eq. 14), 得到具有较高非对映选择性的氢化产物(1R, 2R)-2-羟基环戊烷羧酸乙酯(92%~97% de).研究发现:当催化体系为Ru/49a时, 氢化产物的对映体过量值高达92% ee; 而当配体C4无取代基时(49b), 反应效率最低, 氢化产物的对映体过量值仅为48% ee. Ru/49a催化苯甲酰乙酸乙酯的氢化反应时, 也获得了较高的对映选择性(96% ee).
|
|
(14) |
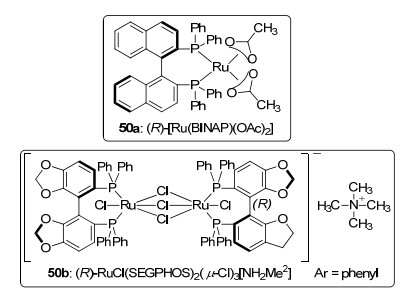
2011年, Floris等[39]报道了几种结构不同的手性钌配合物, 并将它们应用于乙酰乙酸甲酯的不对称氢化反应.研究发现:配体中萘和二唑双膦结构的变化对反应速率和对映选择性影响较大; 具有两个金属中心且有季铵离子特性的催化剂50b [5, 5'-双(二苯基磷)-4, 4'-二-1, 3-苯并二氧杂环, 5, 5'-Bis(diphenylphosphino)-4, 4'-bi-1, 3-benzodioxole, 简称: Segphos], 使氢化产物获得了最高的对映选择性(>99.9% ee); 而当催化剂为50a时, 氢化产物对映体过量值只有6.5%.
2011年, 张兆国课题组[40]以(S)-5, 5'-双(二苯基磷)- 2, 2, 2', 2'-四甲基-4, 4'-二-1, 3-苯并二氧杂环[(S)-SunPhos, 51]为手性配体, 以[Ru(benzene)Cl2]2为催化剂, 催化β-酮酸酯52的不对称氢化反应(Eq. 15).研究发现:与(R)-BINAP 6和C(3)*-TunePhos 45相比, (S)-SunPhos 51的对映选择性更高.底物吸电子氨基保护基可降低分子内氨基相对于酯基的竞争性导向效应, 从而提高氢化产物的对映选择性.使用叔丁氧基羰基保护的底物(52a)时, 目标产物53的对映选择性最高(98.7% ee); 而使用苯甲酰基保护的底物(52b)时, 目标产物53的对映选择性最低(82.4% ee).
|
|
(15) |
同年, 该课题组[41]以Ru/51为催化剂, 催化γ-杂原子取代的β-酮酸酯54的不对称氢化反应(Eq. 16), 得到具有高对映体选择性的β-羟基酯55 (99.1% ee), 并首次通过不对称氢化反应, 获得了具有高对映选择性的4-芳基磺酰基-3-羟基丁酸乙酯55a (97.3% ee).
|
|
(16) |
同年, 该课题组[42]发现以Ru/51为催化剂时, 在EtOH中, β-酮酰胺比β-酮酸酯的氢化反应速率略快; 在四氢呋喃(THF)中, β-酮酰胺的氢化速率比在EtOH中更快, 而酯类的反应速率明显变慢.基于这些发现, 作者将该氢化反应应用于末端由酯和酰胺衍生的3-酮戊二酸衍生物56中(Eq. 17), 得到具有高对映选择性的3-羟基产物57(高达98.1% ee).
|
|
(17) |
2012年, 该课题组[43]在CaCO3存在下, 以Ru/51为催化剂, 催化δ-缩酮-β-酮酸酯58的不对称氢化反应(Eq. 18), 得到具有高对映选择性的δ-缩酮-β-羟基酯59 (>99% ee, 产率79%~97%).研究发现:当未添加CaCO3时, 在58a的氢化反应中未观察到产物产生; 但加入CaCO3后, 可得到具有高对映选择性的氢化产物59a (99.3% ee, 产率79%).作者认为造成这种结果的原因如下: (1) CaCO3在反应中可除去形成催化剂过程中所产生的酸; (2) CaCO3可保持催化剂催化活性.
|
|
(18) |
同年, 该课题组[44]在中性条件下, 以Ru/51为催化剂, 催化γ-卤代-γ, δ-不饱和-β-酮酸酯60的不对称氢化反应(Eq. 19), 得到具有高对映选择性的氢化产物61 (97% ee, 产率98%).在该氢化条件下, β-酮酸酯中C=C和烯基卤素部分在反应过程中保持不变.
|
|
(19) |
同年, 该课题组[45]研究了溶剂对3-酮戊二酸衍生物62不对称氢化反应的影响(Eq. 20).研究发现:在EtOH或THF中, 目标产物63的对映体过量值约为20% ee, 而在丙酮中可增加至90% ee, 当溶剂由EtOH变为THF或丙酮时, 氢化产物63的构型发生反转, 且在混合溶剂中可以得到比在单一溶剂中更高的对映选择性.
|
|
(20) |
2013年, 该课题组[46]以Ru/51为催化剂, 催化β′-酮基-β-氨基酯(64)的不对称氢化反应(Eq. 21), 得到β′-羟基-β-氨基酯(65) (99.6% ee, 98.8% de).研究发现: DCM/2, 2, 2-三氟乙醇(TFE)或DCE/TFE的混合溶液为该反应的最佳溶剂, 且在该混合溶剂体系中, [Ru(cymene)Cl2]2/51表现出比[Ru(cymene)I2]2/51更高的催化活性和对映选择性.
|
|
(21) |
2012年, Satyanarayana等[47]报道了以配合物Ru(H) (p-cymene)((R)-DTBM-Segphos)(SbF6) (66)为催化剂[(R)-(-)-5, 5'-双(二(3, 5-二叔丁基-4-甲氧基苯基)磷)-4, 4'-二-1, 3-苯并二氧烷[(R)-DTBM-Segphos], 催化β′-酮基- β-氨基酯67的不对称氢化反应(Eq. 22), 得到β′-羟基-β-氨基酯68, 该反应在乙醇溶液中进行时, 可获得高对映选择性和较高的非对映选择性(98% ee, 85% de), 与Ru/51[46]催化体系的活性相当.
2013年, Fürstner课题组[48]合成了藻毒素polycave- rnoside A.该合成步骤涉及到β-酮酸酯69的不对称氢化反应(Eq. 23), 该氢化反应以Ru与(R)-MeO-P-Phos 43 (Ar=Ph)形成的配合物为催化剂, 得到相应的氢化产物氯代醇(96% ee, 产率94%), 然后氯代醇中的羟基氢原子被PMB(对甲氧基苄基)取代得到70, 70经一系列反应后则可得到藻毒素polycavernoside A.
|
|
(22) |
|
|
(23) |
2017年, Breit等[49]报道了埃博霉素D的合成, 该合成步骤涉及到3-氧代戊二酸衍生物71a和β, γ-二酮酸酯72b的不对称氢化反应(Eq. 24), 该氢化反应以Ru与(S)-BINAP 7形成的配合物为催化剂, 分别得到具有高对映选择性和高产率的氢化产物72a (99% ee, 产率94%)和79b (86% ee, 产率99%), 72a, 72b经一系列反应后即可得到埃博霉素D.
|
|
(24) |
手性二胺配体来源丰富、易于合成且结构稳定, 在催化不对称合成中表现出良好的对映选择性.
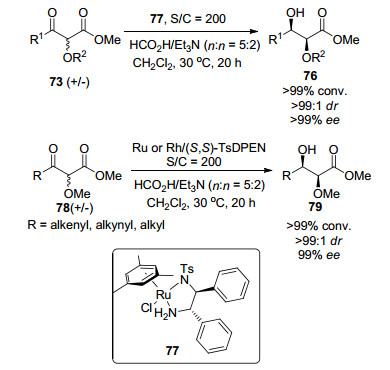
2010年, Ratovelomanana-Vidal等[50]报道了钌催化的α-烷氧基-β-酮酸酯73的不对称转移氢化反应(Eq. 25), 得到具有高非对映选择性和对映选择性的氢化产物70 (99/1 dr, 99% ee).研究表明:该反应最合适的催化剂为RuCl((S, S)-TsDPEN)(η6-arene) (74) [η6-arene=1, 3, 5-三甲基苯, (S, S)-TsDPEN=(S, S)-(-)-2-氨基-1, 2-二苯乙基)(4-甲苯磺酰)氨(75)]; 底物取代基R1和R2的变化对氢化产物76的对映选择性影响不大.
|
|
(25) |
2015年, 该课题组[51]报道了以77为催化剂, 催化(杂)芳基取代的α-烷氧基-β-酮酸酯73不对称转移氢化反应(Scheme 6), 得到具有高非对映选择性和对映选择性的α-烷氧基-β-羟基酯76 (>99:1, dr>99% ee).作者分别以RuⅡ-和RhⅢ-TsDPEN为催化剂, 经进一步研究, 将底物范围扩展到含烯基、炔基和烷基取代基的底物78 (Scheme 6), 得到具有高非对映选择性和对映选择性的α-烷氧基-β-羟基酯79 (>99:1 dr, 99% ee).
同年, 该课题组[52]以Ru/75为催化剂, 催化α-氨基- β-酮酸酯盐酸盐80的不对称转移氢化反应(Eq. 26), 得到具有中等产率(66%~90%)、中等非对映选择性(83:17 dr)和高对映选择性(99:1 er)的氢化产物81.该催化体系的催化活性优于Rh或Ir催化体系.研究发现:杂芳族底物表现出比芳族底物更高的对映体选择性, 但杂芳族底物的非对映体选择性略低.
|
|
(26) |
同年, 该课题组[53]合成了抗生素甲砜霉素的所有立体异构体, 关键步骤为钌催化α-酰胺-β-酮酸酯82的不对称氢化或转移氢化反应, 该反应以Ru/21为催化剂时, 得到具有高非对映选择性和对映选择性的氢化产物83 (>99:1 dr, 95:5 er), 产率为75% (Eq. 27);以Ru/75为催化剂, 甲酸/三乙胺(n:n=5:2, )为氢源时, 氢化产物的产率提高至90%, 且其非对映选择性和对映选择性仍较高(97:3 dr, 97:3 er)(Eq. 28).
|
|
(27) |
2011年, 刘竹青等[54]通过动态动力学拆分的方法, 实现了α-氨基-β-酮酸酯84的不对称转移氢化反应(Eq. 29), 反应以N-((1S, 2R)-2-氨基-1, 2-二苯基乙基)- 2, 3, 4, 5, 6-五氟苯磺酰胺[C6F5SO2DPEN, 85]为手性配体时, 可得到具有高对映体选择性的氢化产物86 (91% ee, 41.3:1 dr), 产率为90%;在相同条件下, 使用其他底物均可获得具有中等产率(80%~99%)和较高对映体选择性(72%~97% ee)的氢化产物; 且该配体的对映选择性明显高于TsDPEN.
|
|
(28) |
|
|
(29) |
2013年, Wills等[55]报道了Ru/(R, R)-TsDPEN催化炔酮和二酮的不对称转移加氢反应, 作者将该反应应用于天然产物(-)-Yashabushidiol B (87)的合成.其中, 当底物为乙炔基取代的β-酮酸酯88时, 可得到对映体过量值高于92% ee的氢化产物89 (Eq. 30), 产率为95%, 89经一系列反应后即可得到(-)-Yashabushidiol B (87).
|
|
(30) |
同年, Mohar等[56]首次报道了具有N, C-(N-乙烯-N-甲基-氨磺酰基)键的(DPEN-κ2N, N′)/η6-arene型钌配合物90, 并将该配合物应用于1-萘基酮的不对称氢化反应中(Eq. 31), 得到具有高对映选择性和高产率的氢化产物(>99.9% ee, 产率100%); 其中, 当底物为β-酮酸酯91时, 氢化产物92对映体过量值高达98.5% ee.
|
|
(31) |
2016年, 该课题组[57]合成了γ-磺内酰胺N, N-配体syn-93和anti-93, 并将该配体应用于钌催化芳基酮的不对称转移氢化反应中(Eq. 32).研究发现:顺式异构体的配体比反式异构体的对映选择性更高, 当底物为β-酮酸酯94a, 94b时, 氢化产物95的对映体过量值大于99% ee.
|
|
(32) |
2017年, 孙国栋等[58]报道了以配合物96为催化剂, 催化芳基α-二苄基氨基-β-酮酸酯97的不对称转移氢化反应(Eq. 33), 得到具有较高非对映选择性和高对映选择性的氢化产物98 (>20:1 de, >99% ee), 产率高达98%.研究发现:底物氮原子上的二苄基取代基不仅在反应的立体化学结果中起关键作用, 而且在温和条件下也容易释放游离氨基而被除去.
|
|
(33) |
手性氨基醇配体的催化效果与它的结构密切相关, 特别是其存在配位能力较强的O、N原子, 可形成多种配合物, 故在不对称催化反应领域占据着重要地位[59].
2010年, Somfai等[60]以(1S, 2S)-2-(苄基氨基)-1, 2-二苯基乙醇[(S, S)-BnDPAE, 99]为手性配体, 以[RuCl2-(benzene)2]为催化剂, 催化α-酰氨基-β-酮酸酯100的不对称转移氢化反应(Eq. 34), 得到具有高对映体选择性和非对映选择性的氢化产物101 (96:4~99:1 er, >95:5 dr), 产率为76%~95%;当底物取代芳基为3-氯苯基时, 氢化产物的对映选择性和产率都较低(66:34 er, 产率69%), 而当底物为3-溴苯基时, 氢化产物的对映选择性和产率却较高(96:4 er, 产率94%).
|
|
(34) |
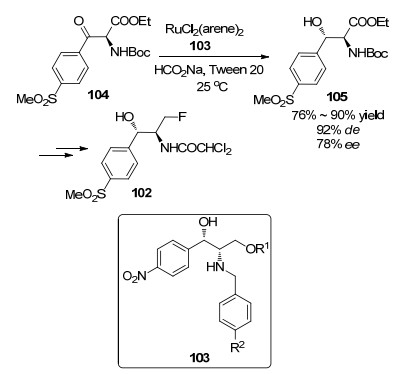
2016年, 陈芬儿等[61]合成了氟苯尼考(102), 该合成的关键步骤是:以103为手性配体, 以RuCl2(arene)2为催化剂, 催化α-氨基-β-酮酸酯104的不对称氢化反应(Scheme 7), 得到具有高非对映选择性和中等对映选择性的(2S, 3S)-α-氨基-β-羟基酯105 (92% de, 78% ee), 产率达76%~90%, 105经一系列反应后即可得到102.
手性P―N配体是一类半稳定的配体, 由于N原子的引入, 配体的电子性质和配位结构发生了较大改变, 表现出了优良的催化活性.
2013年, Kitamura等[62]首次以手性三齿PNN配体2'-(二苯基膦)-N-(吡啶-2-甲基)-(1, 1'-联萘)-2-胺[BINAN-Py-PPh2, 106a]与Ru形成的配合物为催化剂, 催化叔戊酰乙酸甲酯的不对称氢化反应(Eq. 35), 得到具有高对映选择性和高产率的(S)-3-羟基-4, 4-二甲基戊酸甲酯(99:1 er, >99%).研究发现:二甲基亚砜(DMSO)被PyC(6)H…O=S(CH3)2氢键固定到Ru/106a配合物上, 其立体结构, 电子和轨道等因素的协同作用, 可使该氢化反应获得高对映选择性.
|
|
(35) |
2016年, 该课题组[63]将手性PNN配体106a-b应用于酮的加氢反应中(Eq. 36).当底物为107a时, 在甲醇溶剂中, 反应转化率和氢化产物108a的对映选择性都偏低(22%, 85:15 er), 当加入DMSO (1400 mmol•L-1)后, 氢化产物的er值升高为99:1, 转化率大于99%;芳香酮107b在MeOH-DMSO混合溶剂中被氢化, 得到er值为10:90的产物(R)-108b, 转化率只有32%.实验表明: sp3P, sp3N和sp2N原子具有不同的电子性质, DMSO被PyC(6)H…O=S氢键固定到钌配合物上, 且配体中单个sp3NH官能团的存在简化了催化剂反应位点的构建, 这种协同效应使得该不对称氢化反应获得高对映选择性.
|
|
(36) |
20世纪30年代, Lipkin和Stewart[64]就已经将多相金属催化剂引入到不对称催化反应中.与均相催化相比, 多相催化具有催化剂与反应体系易分离、催化剂可循环使用和产物易纯化等特点.多相催化剂主要有以下几种: (1)均相催化剂的多相化, 即将手性配体固定于不溶性载体(通常为聚合物)上, 然后和活性金属配位而成; (2)手性配体修饰的负载型催化剂[65].其中, 实现均相催化剂的多相化有两种方法: (1)将催化剂固定在无机或高分子载体上的固载化均相催化剂; (2)将催化剂动态“担载”在与产物不溶的相来实现两相催化[66].
在多相不对称催化反应中, 固载型催化剂的载体除较常见的SiO2和Al2O3等无机材料外, 还有石墨氧化物、介孔材料(PMOs)、多孔聚合物(CMPs)、金属有机框架材料(MOF)和树状大分子等[67].
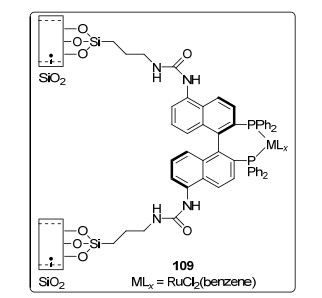
2008年, Koten等[68]报道了SiO2固载化BINAP配体, 并将其分别用于钌和铑催化的烯酰胺, β-酮酸酯和芳香酮的不对称氢化反应.其中, 固载化催化剂109 (图 3), 在乙酰乙酸甲酯和乙酰乙酸乙酯的不对称氢化反应中, 表现出与母体均相催化剂相当的对映选择性(>99% ee), 反应转化率高达100%;该催化剂能催化循环使用5次.
2013年, Park等[69]通过奥古斯丁法, 制备了磷钨酸(PTA)/Al2O3固载化Ru-BINAP催化剂, 并将该催化剂应用于乙酰乙酸甲酯不对称氢化反应中.作者通过用HCl代替乙醇作为浸渍溶剂优化了该方法.研究发现:改性催化剂的催化活性(98.3% ee, 产率95%)优于奥古斯丁催化剂(80.8% ee, 产率14.5%), 且与均相催化剂(99.1% ee, 产率98%)相当, 这是由于酸度的增强而产生的影响; 改性的奥古斯丁催化剂在β-酮酸酯41的不对称氢化反应中, 均表现出较好的催化活性(Eq. 37).
|
|
(37) |
2016年, Kluson等[70]通过直接固定法和杂多酸固定法制备了蒙脱石(硅铝酸盐)固载的Ru/6催化剂, 并将其应用于乙酰乙酸甲酯的不对称氢化反应中.研究发现:当杂多酸为H3PW12O40时, 通过两步法得到的催化剂(MA2N, 97% ee)与均相Ru/6催化剂的催化性能(RUB, 99% ee)相当, 而通过一步法得到的催化剂(MA1N, 94% ee)催化性能稍弱; 当杂多酸为H4SiMo12O40时, 通过一步法得到的催化剂(MSiA1, 53% ee)催化性能明显减弱; 通过直接浸渍固定法得到的催化剂(MONTJAC, 91% ee)也表现出较好的催化性能.
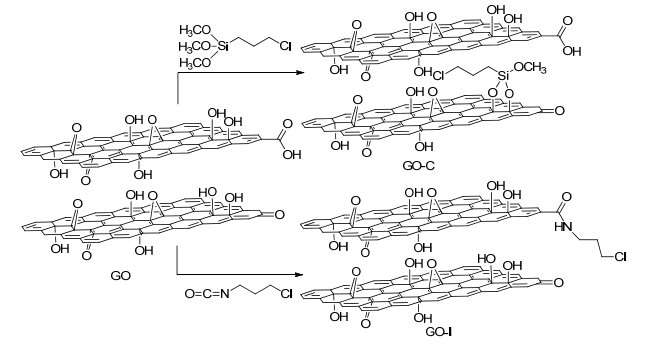
2012年, Bachiller-Baeza等[71]首次研究了石墨氧化物(GO)作为Ru-BINAP催化剂的载体, 他们用烷氧基硅烷或异氰酸酯官能化的GO与手性胺DPEN固定Ru-BINAP, 并将该固载催化剂应用于乙酰乙酸甲酯的不对称氢化反应中(Scheme 8). Ru-GO表现出最高的催化活性(85% ee), 而Ru-GO-C-DPEN (46% ee)和Ru-GO-I-DPEN (45% ee)的催化活性与均相催化剂活性相差较大.该类催化剂在第二次循环使用时, 催化活性都明显下降.
2012年, Crudden等[72]合成了新型双三乙氧基甲硅烷基BINAP单体110, 在表面活性剂存在下, 将110与亚联苯基桥联的硅氧烷前体111共缩合, 得到周期性介孔有机硅(PMOs)官能化的BINAP配体, 并将其应用于β-酮酸酯41的不对称氢化反应(Eq. 38), 得到具有高对映选择性和高产率的氢化产物 (99% ee, 99%).
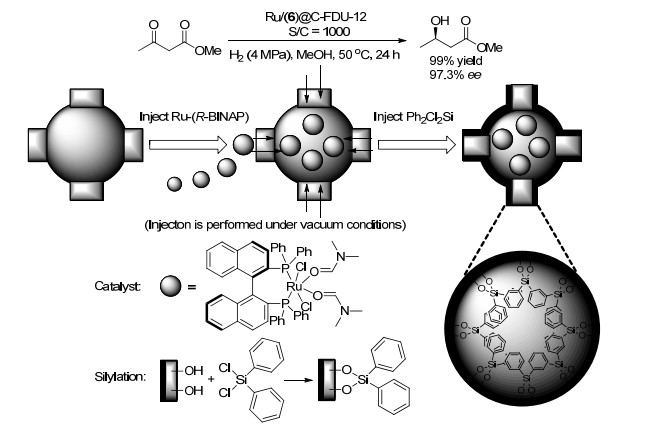
2015年, 杨启华课题组[73]使用活性氯硅烷Ph2Cl2Si作为甲硅烷基化试剂, 将RuCl2(6)(dmf)n封装在介孔二氧化硅C-FDU-12纳米笼中.研究发现: Ru/(6)@C- FDU-12在乙酰乙酸甲酯的不对称氢化反应中(Scheme 9), 表现出较高的催化活性和对映选择性(97.3% ee, 产率99%).分子催化剂与固体载体之间没有强烈的相互作用, 在催化过程中, 被包裹的催化剂可以在纳米反应器中自由移动.该催化剂在一系列β-酮酸酯衍生物的不对称氢化反应中, 均表现出高活性和高对映选择性, 且该催化剂最少能催化循环使用4次.
2012年, 肖丰收等[74]通过二乙烯基苯(DVB)与手性BINAP配体共聚得到PCP-BINAP 112, 112与钌配位形成多相催化剂, 该催化剂在不对称氢化反应中表现出高活性和高对映选择性.当S/C=2000时, β-酮酸酯41在Ru/ PCP-BINAP催化下, 得到具有高对映选择性的氢化产物(94%~99% ee), 转化率大于99.5% (Eq. 51);当S/C达到5000时, 乙酰乙酸甲酯仍能完全转化为相应的氢化产物(90.1% ee), 这是多相催化剂在很高的S/C (5000)比率下, 仍表现出高对映体选择性的第一个实例; 该催化剂最少能催化循环使用6次.
|
|
(38) |
|
|
(39) |
2015年, 李灿课题组[75]合成了一系列含有BINAP配体的手性共轭微孔聚合物BINAP-CMPs, 该聚合物在β-酮酸酯41的不对称氢化反应中(Eq. 40), 表现出高催化活性和高对映体选择性(99% ee, 99% conv.).作者发现, BINAP-CMPs也可应用于铱催化喹哪啶的不对称氢化反应中, 由于该催化剂可阻止二聚体和三聚体的形成, 故表现出比BINAP均相催化剂更高的催化活性.
|
|
(40) |
2017年, 丁云杰课题组[76]合成了(S)-4, 4'-二乙烯基- BINAP和(S)-5, 5′-二乙烯基-BINAP, 并将它们分别与二乙烯基苯共聚合得到BINAP功能化的多孔有机聚合物4-BINAP@POPs和5-BINAP@POPs.在乙酰乙酸甲酯的不对称氢化反应中(Eq. 41), Ru/5-BINAP@POPs-1 (94.3% ee, 产率>99.5%)表现出比Ru/4-BINAP@POPs更高的催化活性; 这可能是由于二乙烯基在4, 4'位比在5, 5'位更接近BINAP的磷原子, 使得Ru/4-BINAP@ POPs催化剂不容易接近底物, 而影响其催化性能.
|
|
(41) |
2018年, 孔胜男等[77]合成了一种手性BINAP功能化的多孔有机聚合物, 该材料具有无定型的微孔结构, 与Ru(Ⅱ)络合形成Ru/POP-BINAP多相催化剂.研究表明:该催化剂在苯甲酰乙酸乙酯的不对称氢化中(Eq. 42), 表现出优良的催化活性和对映选择性(81% ee, 产率98%), 且该催化剂最少能催化循环使用4次.
|
|
(42) |
2014年, 林文斌课题组[78]首次合成了稳定多孔的锆金属-有机框架化合物BINAP-MOF, 并将该化合物分别应用于钌和铑催化的不对称催化反应中.其中, Ru/ BINAP-MOF在β-酮酸酯41的不对称氢化反应中表现出较高的催化活性(Eq. 43), 氢化产物42的对映体过量值高达97% ee; Ru/BINAP-MOF在取代烯烃的氢化反应中同样表现出高催化活性.研究表明: MOF固载化的Ru催化剂与均相Ru催化剂含有相同的Ru配位环境.
|
|
(43) |
2010年, 王伟卫等[79]合成了氟化树枝状手性配体FTsDPEN 113, 并将其应用于前手性酮在水性介质中的不对称转移氢化反应(Eq. 44).当底物为苯甲酰乙酸乙酯时, 得到对映体过量值高达92% ee的氢化产物, 反应转化率大于99%, 该催化剂至少能循环使用26次.
|
|
(44) |
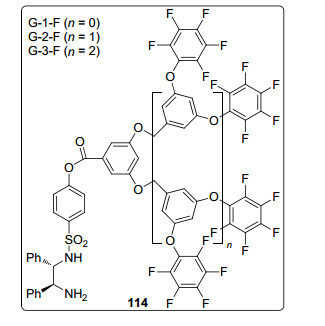
2014年, 该课题组[80]合成了一系列氟化树枝状手性配体114(图 4), 并将其应用于钌催化的不对称氢化反应中.研究发现: Ru/G-2-F在高反应温度(80 ℃)下活性较高, 对于苯甲酰乙酸乙酯的氢化反应, 10 min内转化率可达到99%, 氢化产物的对映体过量值为94% ee; 当反应温度降为40 ℃时, 反应速率减慢, 氢化产物的对映体过量值为92% ee.与传统的非氟化树枝状大分子载体相比, 氟原子的引入可产生分子间弱相互作用(如π-π堆积和氢键相互作用), 使树枝状骨架在催化剂中以半刚性结构存在, 从而提高催化剂的稳定性和循环性能.该催化剂至少能催化循环使用23次.
同年, 范青华课题组[81]合成了一类新型可调树枝状BINAP配体115, 并将该配体分别应用于β-酮酸酯, α-酮酸酯和α-酮酰胺的不对称加氢反应中.研究发现:树枝状大分子的尺寸会影响催化剂的催化性能, 在苯甲酰乙酸甲酯的氢化反应中(Eq. 45), 当催化剂为G0-BINAP和G1-BINAP时, 氢化产物的对映选择性偏低(62% ee, 41% ee), 低于催化剂为MeO-BINAP时的对映选择性(80% ee); 当催化剂为G2-BINAP和G3-BINAP时, 氢化产物3-羟基-3-苯基丙酸甲酯对映选择性明显增加(91% ee, 94% ee).在其他底物的氢化反应中, 均可观察到同样的变化趋势(G3-BINAP>G2-BINAP>MeO- BINAP>G0-BINAP>G1-BINAP).此外, Ru/G2-BINAP催化剂至少能循环使用7次.
|
|
(45) |
在两相体系的不对称催化加氢反应中, 溶剂一般为水、离子液体和超临界CO2等, 这类催化剂反应后与产物处于不相溶的两相, 可通过简单的相分离操作分开, 有着节约原料、降低能耗和对环境友好等优点, 因此具有广阔的发展前景.
2010年, 王伟卫等[82]首次报道了Ru/75在乳液体系中催化酮的不对称转移氢化反应(Eq. 46, 47).研究发现:与在纯水中进行的反应相比, 在乳液中进行的反应可获得更高的转化率和对映选择性(99% ee).例如, 底物为固体酮116a, 116b时, 在乳液体系中目标产物117a, 117b的产率为93%和95%, 对映选择性为53% ee和72% ee; 而该反应以纯水作为溶剂时, 产率(22%, 72%)和对映选择性(43% ee, 71% ee)都降低.作者将底物扩展到各种液体酮, 底物为苯甲酰乙酸乙酯时, 得到了具有高对映选择性的氢化产物(97% ee, 产率>99%).
|
|
(46) |
|
|
(47) |
同年, Zouioueche等[83]分别以酿酒酵母和118a~118c的钌配合物为催化剂, 以水为溶剂, 催化α-酮酸酯和β-酮酸酯的不对称氢化反应(Eq. 48), 作者对这两类催化剂的催化性能进行了比较.研究发现:底物为β-酮酸酯时, 酿酒酵母表现出比Ru/118更高的催化活性.例如, 当底物为乙酰乙酸甲酯时, Ru催化剂的最佳配体为118a, 此时氢化产物的对映体过量值为20% ee, 产率为80%;而当催化剂为酿酒酵母时, 得到对映选择性更高的氢化产物(91.5% ee).
|
|
(48) |
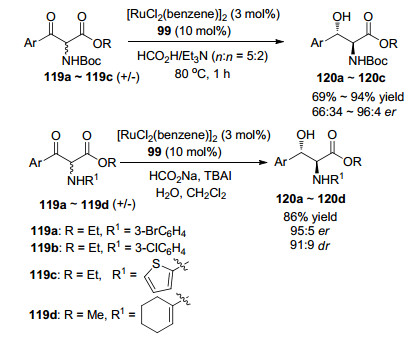
2012年, Somfai等[84]通过两种方法, 实现了α-氨基-β-酮酸酯的不对称转移氢化反应.第一种方法, 以TEAF(HCO2H/Et3N共沸物)为还原剂(Scheme 8), 当底物的取代芳基为3-氯苯基(119b)时, 氢化产物120的对映选择性和产率都较低(66:34 er, 69%), 但底物的取代芳基为3-溴苯基(119a)时, 得到具有高对映选择性和高产率(96:4 er, 94%)的氢化产物120; 而底物为119c时, 对映体选择性和产率都较低(76:24 er, 75%).
第二种方法, 研究了酮在乳液体系中的不对称氢化反应(Scheme 8), 首次通过动态动力学拆分的氢化反应得到了反式烯基β-羟基-α-氨基酯120d.与TEAF法相比, 3-氯底物119b发生反应时, 目标产物120具有更高的对映选择性和产率(96:4 er, 产率85%), 底物119c发生反应时, 目标产物120具有中等对映选择性和非对映选择性(95:5 er, 91:9 dr), 产率为86%.
同年, 该课题组[85]以水为溶剂, 通过动态动力学拆分的不对称转移氢化反应合成了β-羟基-α-(叔丁氧基羰基)-氨基酯122 (Eq. 49).研究发现:由于催化剂和底物具有疏水性, 可通过添加表面活性剂来提高产率.当以RuCl2(benzene)]2与(S, S)-BnDPAE 99形成的催化体系, 催化121的不对称氢化反应时, 在反应中添加中性表面活性剂Tween 20, 可得到具有高对映体选择性的氢化产物122 (97:3 er), 产率大于80%.
|
|
(49) |
2009年, Floris等[86]报道了在甲醇-离子液体(IL)体系中, 以Ru-BINAP为催化剂, 催化乙酰乙酸甲酯的不对称氢化反应.在优化条件下, 以该方式固载的催化剂可以重复使用.作者研究了烷基链在NR222Tf2N离子液体中的作用, 发现反应在N12222Tf2N体系中达到了最高TOF值(509 h-1), 获得了与在纯甲醇系统(98% ee)中相当的对映体选择性(97.5% ee).他们还研究了NR222Br对离子液体的作用, 发现使用N6222Br时, 反应表现出比在纯甲醇系统中更高的对映体选择性(99.1% ee).
同年, Wasserscheid等[87]报道了在甲醇-离子液体体系中, 以Ru/5为催化剂, 催化乙酰乙酸甲酯的不对称氢化反应.研究发现:不同的双(三氟甲基磺酰基)亚胺盐的阳离子对催化剂的催化性能和循环性能有明显影响.当阳离子为羟基烷基季铵离子(123c~123d)时, 在甲醇-离子液体体系中进行的反应获得了与在纯甲醇中相当的对映选择性(95.2% ee), 产率高达98.2%;催化剂在该体系中可循环使用3次.
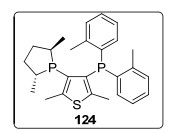
2011年, 该课题组[88]首次报道了负载型离子液体相(SILP)催化剂在连续气相不对称氢化反应中的应用.在离子液体[EMIM][NTf2]中, 二氧化硅30固载化Ru/124催化剂催化乙酰乙酸甲酯时, 氢化产物的对映选择性为65%~82% ee.研究发现, 载体上的离子液体膜对体系的催化活性有明显影响; 通过将Berty反应器改变为管式反应器, 可减少反应中副产物的产生, 并可增强SILP催化剂的稳定性.在反应器中以氦气为载气, 可将底物范围扩展到挥发性较低的物质.
2013年, 金欣等[89]合成了咪唑鎓官能化的BINAP配体125a, 125b, 并将其应用于β-酮酸酯在离子液体(IL)体系中的不对称氢化反应(Eq. 50), Ru/125a, 125b表现出高催化活性和对映选择性(98% ee), 与未修饰的Ru-BINAP催化性能相当.催化剂循环实验表明:在BINAP骨架上引入咪唑基团可显著增强Ru催化剂对IL的亲和力, 减小反应中催化剂的损失, 抑制催化剂氧化, 从而提高催化剂的稳定性和可循环性, 该催化剂可循环使用3次.
|
|
(50) |
2012年, Vinogradov等[90]以Ru/6为催化剂, 在高压CO2下, 催化4-氯-3-氧代丁酸乙酯(69)和乙酰基琥珀酸二甲酯(126)的不对称氢化反应(Eqs. 51, 52).对于底物69, 当反应媒介只有CO2时, 可完全转化为具有高对映选择性的氢化产物127 (92.5% ee); 如果添加[BMIM]BF4作为助溶剂, 氢化产物对映体选择性会增加(96%~97% ee).对于底物126, 需在体系中加入HCl作为助催化剂, 可得到对映体过量值分别为86% ee和28% ee的产物(2R, 3S)-128a和(2R, 3R)-128b; 当反应体系中添加[BMIM]BF4时, 两种产物(2R, 3S)-128a和(2R, 3R)-128的ee值分别提高到90% ee和83% ee.
|
|
(51) |
|
|
(52) |
钌催化β-酮酸酯的不对称氢化反应作为一种高效地合成β-羟基酯的方法已经取得了很大的进展.对于均相不对称催化氢化反应, 可通过改变催化剂配位环境(即配体的类型、配体取代基的电子效应和空间结构)或反应条件来提高其活性和对映选择性.目前, 对此类不对称氢化反应的研究仍在不断地深入, 合成更加高效的配体, 发展立体选择性更高、催化活性强和底物范围更广的钌催化剂仍然是今后的研究热点.
与均相不对称催化氢化相比, 多相不对称催化氢化在分离、操作和催化剂再循环等方面更具优越性, 具有广阔的应用前景.近年来, 人们将均相催化剂与多相催化剂的优点相结合, 实现了均相催化剂的多相化, 并以水、离子液体和超临界液体等作为反应介质, 进一步提高反应体系的选择性, 且符合绿色化学的要求.但到目前为止, 多相不对称催化氢化的研究体系仅局限于为数不多的几个.因此, 优化均相催化剂多相化技术、合成新型固载化催化剂、拓宽催化反应体系、提高催化剂重复使用率是将来需要研究的重点课题之一.
Parshall, G. W.; Nugent, W. A. CHEMTECH 1988, 18, 184.
Parshall, G. W.; Nugent, W. A. CHEMTECH 1988, 18, 376.
殷元骐, 蒋耀忠, 不对称催化反应进展, 科学出版社, 北京, 2000, p. 13.Yin, Y.-Q.; Jiang, Y.-Z. Progress in Asymmetric Catalytic Reactions, Science Press, Beijing, 2000, p. 13(in Chinese).
Knowles, W. S.; Sabacky, M. J. Chem. Commun. 1968, 72, 1445. http://www.ncbi.nlm.nih.gov/pubmed/4270504
Miyashita, A.; Yasuda, A.; Takaya, H.; Toriumi, K.; Ito, T.; Souchi, T.; Noyori, R. J. Am. Chem. Soc. 1980, 102, 7932. doi: 10.1021/ja00547a020
李声时, 自然杂志, 2001, 23, 349. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=ZRZZ200106009&dbname=CJFD&dbcode=CJFQLi, S.-S. Chin. J. Nat. 2001, 23, 349(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=ZRZZ200106009&dbname=CJFD&dbcode=CJFQ
(a) Chen, Q.-A.; Gao, K.; Duan, Y.; Ye, Z.-S.; Shi, L.; Yang, Y.; Zhou, Y.-G. J. Am. Chem. Soc. 2012, 134, 2442.
(b) Li, J.; Shen, J.-F.; Xia, C.; Wang, Y.-Z.; Liu, D.-L.; Zhang, W.-B. Org. Lett. 2016, 18, 2122.
(a) Jiang, J.; Lu, W.-X.; Lv, H.; Zhang, X.-M. Org. Lett. 2015, 17, 1154.
(b) Li, P.; Zhou, M.; Zhao, Q.-Y.; Wu, W.-L.; Hu, X.-Q.; Dong X.-Q.; Zhang, X.-M. Org. Lett. 2016, 18, 40.
(a) Wu, W.-L.; Liu, S.-D.; Duan, M.; Tan, X.-F.; Chen, C.-Y.; Xie, Y.; Lan, Y.; Dong, X.-Q.; Zhang, X.-M. Org. Lett. 2016, 18, 2938.
(b) Wu. W.-L.; You, C.; Yin, C.-C.; Liu, Y.-H.; Dong, X.-Q.; Zhang, X.-M. Org. Lett. 2017, 19, 2548.
(c) Deng, Y.-Y.; Yang, W.; Yang, X.; Yang, D.-Q. Chin. J. Org. Chem. 2017, 37, 3039(in Chinese).
(邓颖颍, 杨文, 杨新, 杨定乔, 有机化学, 2017, 37, 3039.
(a) Mondelli, C.; Vargas, A.; Santarossa, G.; Baiker, A. J. Phys. Chem. C 2009, 113, 15246.
(b) Meemken, F.; Baiker, A.; Dupré, J.; Hungerbühler, K. ACS Catal. 2014, 4, 344.
(c) Guan, S.-L.; Donovan-Sheppard, O.; Reece, C.; Willock, D. J.; Wain, A. J.; Attard, G. A. ACS Catal. 2016, 6, 1822.
(a) Chen, M.-W.; Duan, Y.; Chen, Q.-A.; Wang, D.-S.; Yu, C.-B.; Zhou, Y.-G. Org. Lett. 2010, 12, 5075.
(b) Chen, Z.-P.; Hu, S.-B.; Zhou, J.; Zhou, Y.-G. ACS Catal. 2015, 5, 6086.
吕明, 刘华伟, 广东化工, 2014, 41, 150. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=GDHG201415075&dbname=CJFD&dbcode=CJFQLv, M.; Liu, H.-W. Guangdong Chem. Ind. 2014, 41, 150(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=GDHG201415075&dbname=CJFD&dbcode=CJFQ
Paul, D.; Beiring, B.; Plois, M.; Ortega, N.; Kock, S.; Schlüns, D.; Neugebauer, J.; Wolf, R.; Glorius, F. Organometallics 2016, 35, 3641. doi: 10.1021/acs.organomet.6b00702
Eichenseer, C. M.; Kastl, B.; Pericàs, M. A.; Hanson, P. R.; Reiser, O. ACS Sustainable Chem. Eng. 2016, 4, 2698. doi: 10.1021/acssuschemeng.6b00197
Ding, Z.-Y.; Chen, F.; Qin, J.; He, Y.-M.; Fan, Q.-H. Angew. Chem., Int. Ed. 2012, 51, 5706. doi: 10.1002/anie.201200309
(a) Xie, J.-H.; Zhou, Q.-L. Acta Chim. Sinica 2014, 72, 778(in Chinese).
(谢建华, 周其林, 化学学报, 2014, 72, 778.)
(b) Yuan, Q.-J.; Zhang, W.-B. Chin. J. Org. Chem. 2016, 36, 274(in Chinese).
(袁乾家, 张万斌, 有机化学, 2016, 36, 274.)
郭红超, 丁奎岭, 戴立信, 科学通报, 2004, 49, 1575. doi: 10.3321/j.issn:0023-074X.2004.16.001Guo, H.-C.; Ding, K.-L.; Dai, L.-X. Chin. Sci. Bull. 2004, 49, 1575(in Chinese). doi: 10.3321/j.issn:0023-074X.2004.16.001
Claver, C.; Fernandez, E.; Gillon, A.; Heslop, K.; Hyett, D. J.; Martorell, A.; Orpen, A. G.; Pringle, P. G. Chem. Commun. 2000, 961. doi: 10.1002/chin.200039028/full
Öchsner, E.; Etzold, B.; Junge, K.; Beller, M.; Wasserscheid, P. Adv. Synth. Catal. 2009, 351, 235. doi: 10.1002/adsc.200800531
董建霞, 杨定乔, 刘二畅, 姜凯龄, 韩英峰, 化工时刊, 2005, 19, 40. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=HGJS200503016&dbname=CJFD&dbcode=CJFQDong, J.-X.; Yang, D.-Q.; Liu, E.-C.; Jiang, K.-L.; Han, Y.-F. Chem. Ind. Times 2005, 19, 40(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=HGJS200503016&dbname=CJFD&dbcode=CJFQ
Korostylev, A.; Andrushko, V.; Andrushko, N.; Tararov, V. I.; König, G.; Börner, A. Eur. J. Org. Chem. 2008, 840. http://www.ncbi.nlm.nih.gov/pubmed/19899159
Hamada, Y.; Makino, K. J. Synth. Org. Chem., Jpn. 2008, 66, 1057. doi: 10.5059/yukigoseikyokaishi.66.1057
Makino, K.; Goto, T.; Hiroki, Y.; Hamada, Y. Tetrahedron:Asymmetry 2008, 19, 2816. doi: 10.1016/j.tetasy.2008.12.024
Makino, K.; Goto, T.; Ohtaka, J.; Hamada, Y. Heterocycles 2009, 77, 629. doi: 10.3987/COM-08-S(F)46
Roche, C.; Desroy, N.; Haddad, M.; Phansavath, P.; Genêt, J. P. Org. Lett. 2008, 10, 3911. doi: 10.1021/ol801493w
Roche, C.; Roux, R. L.; Haddad, M.; Phansavath, P.; Genêt, J. P. Synlett 2009, 573.
Prévost, S.; Gauthier, S.; De Andrade, M. C.; Mordant, C.; Touati, A. R.; Lesot, P.; Savignac, P.; Ayad, T.; Phansavath, P.; Ratovelomanana-Vidal, V.; Genêt, J. P. Tetrahedron:Asymmetry 2010, 21, 1436. doi: 10.1016/j.tetasy.2010.05.017
Prévost, S.; Ayad, T.; Phansavath, P.; Ratovelomanana-Vidal, V. Adv. Synth. Catal. 2011, 353, 3213. doi: 10.1002/adsc.v353.17
Touati, R.; Hassine, B. B. J. Soc. Chim. Tunis. 2008, 10, 127.
Kesselgruber, M.; Lotz, M.; Martin, P.; Melone, G.; Müller, M.; Pugin, B.; Naud, F.; Spindler, F.; Thommen, M.; Zbinden, P.; Blaser, H. U. Chem. Asian J. 2008, 3, 1384. doi: 10.1002/asia.v3:8/9
陈丽, 马梦林, 彭宗海, 陈华, 有机化学, 2008, 28, 1724. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract338090.shtmlChen, L.; Ma, M.-L.; Peng, Z.-H.; Chen, H. Chin. J. Org. Chem. 2008, 28, 1724(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract338090.shtml
彭宗海, 马梦林, 付海燕, 陈华, 李贤钧, 催化学报, 2010, 31, 191. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=CHUA201002013&dbname=CJFD&dbcode=CJFQPeng, Z.-H.; Ma, M.-L.; Fu, H.-Y.; Chen, H.; Li, X.-J. Chin. J. Catal. 2010, 31, 191(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=CHUA201002013&dbname=CJFD&dbcode=CJFQ
Sun, X.-F.; Li, W.; Hou, G.-H.; Zhou, L.; Zhang, X.-M. Adv. Synth. Catal. 2009, 351, 2553. doi: 10.1002/adsc.200900589
Wang, C.-J.; Wang, C.-B.; Chen, D.; Yang, G.-F.; Wu, Z.-S.; Zhang, X.-M. Tetrahedron Lett. 2009, 50, 1038. doi: 10.1016/j.tetlet.2008.12.057
Zhang, Z.; Qian, H.; Longmire, J.; Zhang, X. J. Org. Chem. 2000, 65, 6223. doi: 10.1021/jo000462v
Sun, X.-F.; Li, W.; Zhou, L.; Zhang, X.-M. Chem.-Eur. J. 2009, 15, 7302. doi: 10.1002/chem.v15:30
Yuan, W.-C.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. Tetrahedron 2009, 65, 4130. doi: 10.1016/j.tet.2009.03.066
Oki, H.; Oura, I.; Nakamura, T.; Ogata, K.; Fukuzawa, S. I. Tetrahedron:Asymmetry 2009, 20, 2185. doi: 10.1016/j.tetasy.2009.09.004
Floris, T.; Kluson, P.; Slater, M. React. Kinet., Mech. Catal. 2011, 102, 67. doi: 10.1007/s11144-010-0256-1
Yao, Y.; Fan, W.-Z.; Li, W.-F.; Ma, X.; Zhu, L.-F.; Xie, X.-M.; Zhang, Z.-G. J. Org. Chem. 2011, 76, 2807. doi: 10.1021/jo2002165
Fan, W.-Z.; Li, W.-F.; Ma, X.; Tao, X.-M.; Li, X.-M.; Yao, Y.; Xie, X.-M.; Zhang, Z.-G. J. Org. Chem. 2011, 76, 9444. doi: 10.1021/jo201822k
Li, W.-F.; Ma, X.; Fan, W.-Z.; Tao, X.-M.; Li, X.-M.; Xie, X.-M.; Zhang, Z.-G. Org. Lett. 2011, 13, 3876. doi: 10.1021/ol201406w
Fan, W.-Z.; Li, W.-F.; Ma, X.; Tao, X.-M.; Li, X.-M.; Yao, Y.; Xie, X.-M.; Zhang, Z.-G. Chem. Commun. 2012, 48, 4247. doi: 10.1039/c2cc31002c
Ma, X.; Li, W.-F.; Li, X.-M.; Tao, X.-M.; Fan, W.-Z.; Xie, X.-M.; Ayad, T.; Ratovelomanana-Vidal, V.; Zhang, Z.-G. Chem. Commun. 2012, 48, 5352. doi: 10.1039/c2cc30925d
Li, W.-F.; Tao, X.-M.; Ma, X.; Fan, W.-Z.; Li, X.-M.; Zhao, M.-M.; Xie, X.-M.; Zhang, Z.-G. Chem.-Eur. J. 2012, 18, 16531. doi: 10.1002/chem.201202614
Li, X.-M.; Tao, X.-M.; Ma, X.; Li, W.-F.; Zhao, M.-M.; Xie, X.-M.; Ayad, T.; Ratovelomanana-Vidal, V.; Zhang, Z.-G. Tetrahedron 2013, 69, 7152. doi: 10.1016/j.tet.2013.05.136
Satyanarayana, P.; Maheswaran, H.; Kantama, M. L.; Chawla, H. P. S. Catal. Sci. Technol. 2012, 2, 2508. doi: 10.1039/c2cy20335a
Brewitz, L.; Llaveria, J.; Yada, A.; Fürstner, A. Chem.-Eur. J. 2013, 19, 4532. doi: 10.1002/chem.201204551
Haydl, A. M.; Breit, B. Chem.-Eur. J. 2017, 23, 541. doi: 10.1002/chem.201605011
Cartigny, D.; Püntener, K.; Ayad, T.; Scalone, M.; Ratovelomanana-Vidal, V. Org. Lett. 2010, 12, 3788. doi: 10.1021/ol101451s
Monnereau, L.; Cartigny, D.; Scalone, M.; Ayad, T.; Ratovelomanana-Vida, V. Chem.-Eur. J. 2015, 21, 11799. doi: 10.1002/chem.v21.33
Echeverria, P. G.; Cornil, J.; Férard, C.; Guérinot, A.; Cossy, J.; Phansavath, P.; Ratovelomanana-Vidal, V. RSC Adv. 2015, 5, 56815. doi: 10.1039/C5RA10385A
Perez, M.; Echeverria, P. G.; Martinez-Arripe, E.; Zoubir, M. E.; Touati, R.; Zhang, Z.-G.; Genet, J. P.; Phansavath, P.; Ayad, T.; Ratovelomanana-Vidal, V. Eur. J. Org. Chem. 2015, 5949. doi: 10.1002/ejoc.201590076/pdf
Liu, Z.-Q.; Shultz, C. S.; Sherwood, C. A.; Krska, S.; Dormer, P. G.; Desmond, R.; Lee, C.; Sherer, E. C.; Shpungin, J.; Cuff, J.; Xu, F. Tetrahedron Lett. 2011, 52, 1685. doi: 10.1016/j.tetlet.2011.01.146
Fang, Z.-J.; Wills, M. J. Org. Chem. 2013, 78, 8594. doi: 10.1021/jo401284c
Kišić, A.; Stephan, M.; Mohar, B. Org. Lett. 2013, 15, 1614. doi: 10.1021/ol400393j
Rast, S.; Modec, B.; Stephan, M.; Mohar, B. Org. Biomol. Chem. 2016, 14, 2112. doi: 10.1039/C5OB02352A
Sun, G.-D.; Zhou, Z.-H.; Luo, Z.-H.; Wang, H.-L.; Chen, L.; Xu, Y.-B.; Li, S.; Jian, W.-L.; Zeng, J.-B.; Hu, B.-Q.; Han, X.-D.; Lin, Y.-C.; Wang, Z.-Q. Org. Lett. 2017, 19, 4339. doi: 10.1021/acs.orglett.7b01982
翁文, 周宏英, 傅宏祥, 吕士杰, 有机化学, 1998, 18, 509. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract326158.shtmlWeng, W.; Zhou, H.-Y.; Fu, H.-X.; Lv, S.-J. Chin. J. Org. Chem. 1998, 18, 509(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract326158.shtml
Seashore-Ludlow, B.; Villo, P.; Häcker, C.; Somfai, P. Org. Lett. 2010, 12, 5274. doi: 10.1021/ol102323k
Wang, X.-L.; Xu, L.-J.; Yan, L.-J.; Wang, H.-F.; Han, S.; Wu, Y.; Chen, F.-E. Tetrahedron 2016, 72, 1787. doi: 10.1016/j.tet.2016.02.045
Yamamura, T.; Nakatsuka, H.; Tanaka, S.; Kitamura, M. Angew. Chem. 2013, 125, 9483. doi: 10.1002/ange.201304408
Yamamura, T.; Nakane, S.; Nomura, Y.; Tanaka, S.; Kitamura, M. Tetrahedron 2016, 72, 3781. doi: 10.1016/j.tet.2016.02.007
Lipkin, D.; Stewart, T. D. J. Am. Chem. Soc. 1939, 61, 3297. doi: 10.1021/ja01267a017
左晓斌, 刘汉范, 分子催化, 1997, 11, 309. http://www.cnki.com.cn/Article/CJFD1997-FZCH704.014.htmZuo, X.-B.; Liu, H.-F. J. Mol. Catal. 1997, 11, 309(in Chinese). http://www.cnki.com.cn/Article/CJFD1997-FZCH704.014.htm
鲁福身, 晁建平, 杨春育, 化学进展, 2001, 13, 192. doi: 10.3321/j.issn:1005-281X.2001.03.005Lu, F.-S.; Chao, J.-P.; Yang, C.-Y. Prog. Chem. 2001, 13, 192(in Chinese). doi: 10.3321/j.issn:1005-281X.2001.03.005
Song, C. E. Annu. Rep. Prog. Chem., Sect. C:Phys. Chem. 2005, 101, 143. doi: 10.1039/b408828j
McDonald, A. R.; Müller, C.; Vogt, D.; Klinka, G. P. M. V.; Koten, G. V. Green Chem. 2008, 10, 424. doi: 10.1039/b714189k
Ahn, S. H.; Choi, M. S.; Im, J. S.; Sheikh, R.; Park, Y. H. J. Mol. Catal. A:Chem. 2013, 373, 55. doi: 10.1016/j.molcata.2013.03.001
Kluson, P.; Krystynik, P.; Dytrych, P.; Bartek, L. React. Kinet., Mech. Cat. 2016, 119, 393. doi: 10.1007/s11144-016-1078-6
Dongil, A. B. Bachiller-Baeza, B.; Guerrero-Ruiz, A.; Rodríguez-Ramos, I. Catal. Commun. 2012, 26, 149. doi: 10.1016/j.catcom.2012.05.015
Seki, T.; McEleney, K.; Crudden, C. M. Chem. Commun. 2012, 48, 6369. doi: 10.1039/c2cc31247f
Peng, J.; Wang, X.-F.; Zhang, X.-M.; Bai, S.-Y.; Zhao, Y.-P.; Li, C.; Yang, Q.-H. Catal. Sci. Technol. 2015, 5, 666. doi: 10.1039/C4CY00228H
Sun, Q.; Meng, X.-J.; Liu, X.; Zhang, X.-M.; Yang, Y.; Yang, Q.-H.; Xiao, F.-S. Chem. Commun. 2012, 48, 10505. doi: 10.1039/c2cc35192g
Wang, X.; Lu, S.-M.; Li, J.; Liu, Y.; Li, C. Catal. Sci. Technol. 2015, 5, 2585. doi: 10.1039/C5CY00038F
Wang, T.; Lyu, Y.; Xiong, K.; Wang, W.-L.; Zhang, H.; Zhan, Z.-P.; Jiang, Z.; Ding, Y.-J. Chin. J. Catal. 2017, 38, 890. doi: 10.1016/S1872-2067(17)62826-2
孔胜男, Abaid Ullah Malik, 钱雪峰, 舒谋海, 肖文德, 有机化学, 2018, 38, 656. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346360.shtmlKong, S.-N.; Malik, A. U.; Qian, X.-F.; Shu, M.-H.; Xiao, W.-D. Chin. J. Org. Chem. 2018, 38, 656(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346360.shtml
Falkowski, J. M.; Sawano, T.; Zhang, T.; Tsun, G.; Chen, Y.; Lockard, J. V.; Lin, W.-B. J. Am. Chem. Soc. 2014, 136, 5213. doi: 10.1021/ja500090y
Wang, W.-W.; Wang, Q.-R. Chem. Commun. 2010, 46, 4616. doi: 10.1039/c002168g
Wang, W.-W.; Li, Z.-M.; Su, L.; Wang, Q.-R.; Wu, Y.-L. J. Mol. Catal., A 2014, 387, 92. doi: 10.1016/j.molcata.2014.02.030
Ma, B.-D.; Miao, T.-T.; Sun, Y.-H.; He, Y.-M.; Liu, J.; Feng, Y.; Chen, H.; Fan, Q.-H. Chem.-Eur. J. 2014, 20, 9969. doi: 10.1002/chem.201402709
Wang, W.-W.; Li, Z.-M.; Mu, W.-B.; Su, L.; Wang, Q.-R. Catal. Commun. 2010, 11, 480. doi: 10.1016/j.catcom.2009.12.002
Zeror, S.; Collin, J.; Fiaud, J. C.; Zouioueche, L. A. Tetrahedron:Asymmetry 2010, 21, 1211. doi: 10.1016/j.tetasy.2010.05.014
Seashore-Ludlow, B.; Villo, P.; Somfai, P. Chem.-Eur. J. 2012, 18, 7219. doi: 10.1002/chem.201103739
Seashore-Ludlow, B. Saint-Dizier, F.; Somfai, P. Org. Lett. 2012, 14, 6334. doi: 10.1021/ol303115v
Floris, T.; Kluson, P.; Bartek, L.; Pelantova, H. Appl. Catal., A 2009, 366, 160. doi: 10.1016/j.apcata.2009.07.002
Öchsner, E.; Schneiders, K.; Junge, K.; Beller, M.; Wasserscheid, P. Appl. Catal., A 2009, 364, 8. doi: 10.1016/j.apcata.2009.05.020
Öchsner, E.; Schneider, M. J.; Meyer, C.; Haumann, M.; Wasserscheid, P. Appl. Catal., A 2011, 399, 35. doi: 10.1016/j.apcata.2011.03.038
Jin, X.; Kong, F.-F.; Yang, Z.-Q.; Cui, F.-F. J. Mol. Catal. A-Chem. 2013, 374, 22. http://www.sciencedirect.com/science/article/pii/S1381116913001167
Turova, O. V.; Kuchurov, I. V.; Starodubtseva, E. V.; Ferapontov, V. A.; Ikonnikov, N. S.; Zlotina, S. G.; Vinogradov, M. G. Mendeleev Commun. 2012, 22, 184. doi: 10.1016/j.mencom.2012.06.003

图式 1 Ru催化α-氨基-β-酮酸酯不对称氢化反应可能的机理
Scheme 1 Plausible mechanism for Ru-catalyed asymmetric hydrogenation of α-amino-β-ketoesters

图式 2 (2R, 3R)-2-氨基-3-环己基-3-羟基丙酸的合成
Scheme 2 Synthesis of (2R, 3R)-2-amino-3-cyclohexyl-3- hydroxypropionic acid

图式 3 Dolabelide A C15~C30片段的合成
Scheme 3 Synthesis of the C15~C30 subunit of dolabelide A

图式 4 钌催化β-酮酸酯不对称氢化
Scheme 4 Ruthenium-catalyzed asymmetric hydrogenation of β-keto esters

图式 5 4, 4'-二叔丁基TunePhos型手性双膦配体在不对称氢化中的应用
Scheme 5 Application of 4, 4'-di-tert-butyl TunePhos type chiral diphosphine ligand in asymmetric hydrogenation

图式 6 Ru或Rh催化β-酮酸酯不对称氢化
Scheme 6 Ru- or Rh-catalyzed asymmetric hydrogenation of β-keto esters

图 2 用于钌催化不对称氢化反应的双膦配体
Figure 2 Diphosphine ligands for ruthenium-catalyzed asymmetric hydrogenation

图式 8 有机硅烷和异氰酸酯官能化的GO
Scheme 8 Functionalization of GO with organosilane (GO-C) and isocyanate (GO-I)

图式 9 将Ru/6封装在C-FDU-12纳米笼中的示意图
Scheme 9 Schematic illustration of encapsulating Ru-6 in the nanocage of C-FDU-12

图式 8 α-氨基-β-酮酸酯的不对称转移氢化
Scheme 8 Asymmetric transfer hydrogenation of α-amino-β- keto ester

图 6 NR222Tf2N型离子对的分子结构
Figure 6 Molecular structures of the ionic pairs of the NR222Tf2N type of IL

图 7 双(三氟甲基磺酰基)酰亚胺离子液体的阳离子
Figure 7 Cations of the bis(trifluoromethylsulfonyl)imide ionic liquids

图 8 用于连续气相不对称氢化反应的配体
Figure 8 Ligand for continuous gas phase asymmetric hydrogenation
表 1 杂化配体氢化β-酮酸酯
Table 1. Hydrogenations of β-ketoester with hybrid ligands
|
|
||||
| Entry | L* | Conv./% | ee/% | Config. |
| 1 | |
100 | 78 | R |
| 2 | |
100 | 83 | R |
| 3 | |
100 | 89 | R |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们