-
[1]
Feng D, Lei T, Lukatskaya M R, et al. Nat. Energy, 2018, 3(1): 30~36. doi: 10.1038/s41560-017-0044-5
-
[2]
Wu H B, Lou X W. Sci. Adv., 2017, 3(12): eaap9252.
-
[3]
Yang C, Schellhammer K S, Ortmann F, et al. Angew. Chem. Int. Ed., 2017, 56(14): 3920~3924. doi: 10.1002/anie.201700679
-
[4]
Li S L, Xu Q. Energy Environ. Sci., 2013, 6(6): 1656~1683. doi: 10.1039/c3ee40507a
-
[5]
Rogge S M J, Bavykina A, Hajek J, et al. Chem. Soc. Rev., 2017, 46(11): 3134~3184. doi: 10.1039/C7CS00033B
-
[6]
Xu H, Gao J, Jiang D. Nat. Chem., 2015, 7(11): 905~912. doi: 10.1038/nchem.2352
-
[7]
Duan J, Chen S, Zhao C. Nat. Commun., 2017, 8: 15341. doi: 10.1038/ncomms15341
-
[8]
Lee J, Farha O K, Roberts J, et al. Chem. Soc. Rev., 2009, 38(5): 1450~1459. doi: 10.1039/b807080f
-
[9]
Chughtai A H, Ahmad N, Younus H A, et al. Chem. Soc. Rev., 2015, 44(19): 6804~6849. doi: 10.1039/C4CS00395K
-
[10]
Lu M, Liu J, Li Q, et al. Angew. Chem. Int. Ed., 2019, 58(36): 12392~12397. doi: 10.1002/anie.201906890
-
[11]
Zhao S, Wang Y, Dong J, et al. Nat. Energy, 2016, 1(12): 16184. doi: 10.1038/nenergy.2016.184
-
[12]
Zhao H, Jin Z, Su H, et al. Chem. Commun., 2011, 47(22): 6389~6391. doi: 10.1039/c1cc00084e
-
[13]
Horcajada P, Chalati T, Serre C, et al. Nat. Mater., 2010, 9(2): 172~178. doi: 10.1038/nmat2608
-
[14]
Peng Y, Li Y, Ban Y, et al. Angew. Chem. Int. Ed., 2017, 56(33): 9757~9761. doi: 10.1002/anie.201703959
-
[15]
Rodenas T, Luz I, Prieto G, et al. Nat. Mater., 2015, 14(1): 48~55. doi: 10.1038/nmat4113
-
[16]
Li J R, Kuppler R J, Zhou H C. Chem. Soc. Rev., 2009, 38(5): 1477~1504. doi: 10.1039/b802426j
-
[17]
Doonan C J, Tranchemontagne D J, Glover T G, et al. Nat. Chem., 2010, 2(3): 235~238. doi: 10.1038/nchem.548
-
[18]
Getman R B, Bae Y S, Wilmer C E, et al. Chem. Rev., 2012, 112(2): 703~723. doi: 10.1021/cr200217c
-
[19]
Huang N, Ding X, Kim J, et al. Angew. Chem. Int. Ed., 2015, 54(30): 8704~8707. doi: 10.1002/anie.201503902
-
[20]
Wang M, Ballabio M, Wang M, et al. J. Am. Chem. Soc., 2019, 141(42): 16810~16816. doi: 10.1021/jacs.9b07644
-
[21]
Xu H, Tao S, Jiang D. Nat. Mater., 2016, 15(7): 722~726. doi: 10.1038/nmat4611
-
[22]
Campbell M G, Sheberla D, Liu S F, et al. Angew. Chem. Int. Ed., 2015, 54(14): 4349~4352. doi: 10.1002/anie.201411854
-
[23]
Zhu J, Yang C, Lu C, et al. Acc. Chem. Res., 2018, 51(12): 3191~3202. doi: 10.1021/acs.accounts.8b00444
-
[24]
Novoselov K S, Geim A K, Morozov S V, et al. Science, 2004, 306(5696): 666. doi: 10.1126/science.1102896
-
[25]
Côté A P, Benin A I, Ockwig N W, et al. Science, 2005, 310: 1166~1170. doi: 10.1126/science.1120411
-
[26]
Jin E, Asada M, Xu Q, et al. Science, 2017, 357(6352): 673~676. doi: 10.1126/science.aan0202
-
[27]
Kuhn P, Antonietti M, Thomas A. Angew. Chem. Int. Ed., 2008, 47(18): 3450~3453. doi: 10.1002/anie.200705710
-
[28]
Diercks C S, Yaghi O M. Science, 2017, 355(6328): eaal1585.
-
[29]
Spitler E L, Dichtel W R. Nat. Chem., 2010, 2(8): 672~677. doi: 10.1038/nchem.695
-
[30]
Feng X, Ding X, Jiang D. Chem. Soc. Rev., 2012, 41(18): 6010~6022. doi: 10.1039/c2cs35157a
-
[31]
Li H, Eddaoudi M, O'Keeffe M, et al. Nature, 1999, 402(6759): 276~279. doi: 10.1038/46248
-
[32]
Dong R, Han P, Arora H, et al. Nat. Mater., 2018, 17(11): 1027~1032. doi: 10.1038/s41563-018-0189-z
-
[33]
Zhao M, Wang Y, Ma Q, et al. Adv. Mater., 2015, 27(45): 7372~7378. doi: 10.1002/adma.201503648
-
[34]
Zhou H C, Kitagawa S. Chem. Soc. Rev., 2014, 43(16): 5415~5418. doi: 10.1039/C4CS90059F
-
[35]
Furukawa H, Cordova K E, O'Keeffe M, et al. Science, 2013, 341(6149): 1230444. doi: 10.1126/science.1230444
-
[36]
Colson J W, Woll A R, Mukherjee A, et al. Science, 2011, 332(6026): 228~231. doi: 10.1126/science.1202747
-
[37]
Lin S, Diercks C S, Zhang Y B, et al. Science, 2015, 349: 1208~1213. doi: 10.1126/science.aac8343
-
[38]
Zhang B, Mao H, Matheu R, et al. J. Am. Chem. Soc., 2019, 141(29): 11420~11424. doi: 10.1021/jacs.9b05626
-
[39]
Jiang J X, Su F, Trewin A, et al. J. Am. Chem. Soc., 2008, 130(24): 7710~7720. doi: 10.1021/ja8010176
-
[40]
Bi S, Yang C, Zhang W, et al. Nat. Commun., 2019, 10(1): 2467. doi: 10.1038/s41467-019-10504-6
-
[41]
Zhuang X, Zhao W, Zhang F, et al. Polym. Chem., 2016, 7(25): 4176~4181. doi: 10.1039/C6PY00561F
-
[42]
Zheng C, Zhu J, Yang C, et al. Sci. China Chem., 2019, 62(9): 1145~1193. doi: 10.1007/s11426-019-9477-y
-
[43]
Yuan Y, Yang Y, Ma X, et al. Adv. Mater., 2018, 30(12): e1706507.
-
[44]
Chen L, Yang Y, Jiang D. J. Am. Chem. Soc., 2010, 132(26): 9138~9143. doi: 10.1021/ja1028556
-
[45]
Dong J, Zhang K, Li X, et al. Nat. Commun., 2017, 8(1): 1142. doi: 10.1038/s41467-017-01293-x
-
[46]
Zhang T, Qi H, Liao Z, et al. Nat. Commun., 2019, 10(1): 4225. doi: 10.1038/s41467-019-11921-3
-
[47]
Xu S, Wang G, Biswal B P, et al. Angew. Chem. Int. Ed., 2019, 58(3): 849~853. doi: 10.1002/anie.201812685
-
[48]
Liu W, Luo X, Bao Y, et al. Nat. Chem., 2017, 9(6): 563~570. doi: 10.1038/nchem.2696
-
[49]
Kissel P, Erni R, Schweizer W B, et al. Nat. Chem., 2012, 4(4): 287~291. doi: 10.1038/nchem.1265
-
[50]
Kory M J, Worle M, Weber T, et al. Nat. Chem., 2014, 6(9): 779~784. doi: 10.1038/nchem.2007
-
[51]
Su Y, Yao Z, Zhang F, et al. Adv. Funct. Mater., 2016, 26(32): 5893~5902. doi: 10.1002/adfm.201602158
-
[52]
Liu K, Qi H, Dong R, et al. Nat. Chem., 2019, 11(11): 994~1000. doi: 10.1038/s41557-019-0327-5
-
[53]
Liu X, Xu Y, Jiang D. J. Am. Chem. Soc., 2012, 134(21): 8738~8741. doi: 10.1021/ja303448r
-
[54]
Chen L, Honsho Y, Seki S, et al. J. Am. Chem. Soc., 2010, 132(19): 6742~6748. doi: 10.1021/ja100327h
-
[55]
Franc G, Gourdon A. Phys. Chem. Chem. Phys., 2011, 13(32): 14283~14292. doi: 10.1039/c1cp20700h
-
[56]
Ruffieux P, Wang S, Yang B, et al. Nature, 2016, 531(7595): 489~492. doi: 10.1038/nature17151
-
[57]
Rizzo D J, Veber G, Cao T, et al. Nature, 2018, 560(7717): 204~208. doi: 10.1038/s41586-018-0376-8
-
[58]
Li C J. Acc. Chem. Res., 2008, 42(2): 335~344.
-
[59]
Zhang X, Zeng Q, Wang C. Nanoscale, 2013, 5(18): 8269~8287. doi: 10.1039/c3nr01611k
-
[60]
Zhuang X, Mai Y, Wu D, et al. Adv. Mater., 2015, 27(3): 403~427. doi: 10.1002/adma.201401857
-
[61]
Bielecki F U J. Ber. Deutschen Chem. Gesel., 1901, 34: 2174~2185. doi: 10.1002/cber.190103402141
-
[62]
Ullmann F. Jus. Lieb. Ann. Chem., 1904, 332: 38~81. doi: 10.1002/jlac.19043320104
-
[63]
Judd C J, Haddow S L, Champness N R, et al. Sci. Rep., 2017, 7(1): 14541. doi: 10.1038/s41598-017-13315-1
-
[64]
Grill L, Dyer M, Lafferentz L, et al. Nat. Nanotech., 2007, 2(11): 687~691. doi: 10.1038/nnano.2007.346
-
[65]
Lafferentz L, Eberhardt V, Dri C, et al. Nat. Chem., 2012, 4(3): 215~220. doi: 10.1038/nchem.1242
-
[66]
Morchutt C, Bjork J, Krotzky S, et al. Chem. Commun., 2015, 51(12): 2440~2443. doi: 10.1039/C4CC07107G
-
[67]
Gutzler R, Walch H, Eder G, et al. Chem. Commun., 2009, 29(29): 4456~4458.
-
[68]
Blunt M O, Russell J C, Champness N R, et al. Chem. Commun., 2010, 46(38): 7157~7159. doi: 10.1039/c0cc01810d
-
[69]
Fritton M, Duncan D A, Deimel P S, et al. J. Am. Chem. Soc., 2019, 141(12): 4824~4832. doi: 10.1021/jacs.8b11473
-
[70]
Eichhorn J, Nieckarz D, Ochs O, et al. ACS Nano, 2014, 8(8): 7880~7889. doi: 10.1021/nn501567p
-
[71]
Bieri M, Treier M, Cai J, et al. Chem. Commun., 2009, (45): 6919~6921. doi: 10.1039/b915190g
-
[72]
Bieri M, Nguyen M T, Gröning O, et al. J. Am. Chem. Soc., 2010, 132(46): 16669~16676. doi: 10.1021/ja107947z
-
[73]
Cardenas L, Gutzler R, Lipton-Duffin J, et al. Chem. Sci., 2013, 4(8): 3263~3268. doi: 10.1039/c3sc50800e
-
[74]
Smykalla L, Shukrynau P, Korb M, et al. Nanoscale, 2015, 7(9): 4234~4241. doi: 10.1039/C4NR06371F
-
[75]
Mondal S. ChemTexts, 2016, 2(4): 17. doi: 10.1007/s40828-016-0036-2
-
[76]
Reppe W, Schlichting O, Klager K, et al. Jus. Lieb. Ann. Chem., 1948, 560(1): 1~92. doi: 10.1002/jlac.19485600102
-
[77]
Liu J, Ruffieux P, Feng X, et al. Chem. Commun., 2014, 50(76): 11200~11203. doi: 10.1039/C4CC02859G
-
[78]
Zhou H, Liu J, Du S, et al. J. Am. Chem. Soc., 2014, 136(15): 5567~5570. doi: 10.1021/ja501308s
-
[79]
Yang B, Bjork J, Lin H, et al. J. Am. Chem. Soc., 2015, 137(15): 4904~4907. doi: 10.1021/jacs.5b00774
-
[80]
Scheuermann C J. Chem. Asian J., 2010, 5(3): 436~451. doi: 10.1002/asia.200900487
-
[81]
Sun Q, Zhang C, Cai L, et al. Chem. Commun., 2015, 51(14): 2836~2839. doi: 10.1039/C4CC08299K
-
[82]
Li Q, Yang B, Lin H, et al. J. Am. Chem. Soc., 2016, 138(8): 2809~2814. doi: 10.1021/jacs.5b13286
-
[83]
Abel M, Clair S, Ourdjini O, et al. J. Am. Chem. Soc., 2011, 133(5): 1203~1205. doi: 10.1021/ja108628r

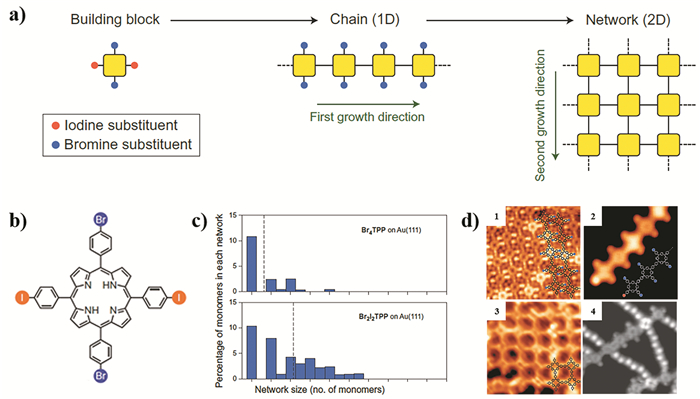
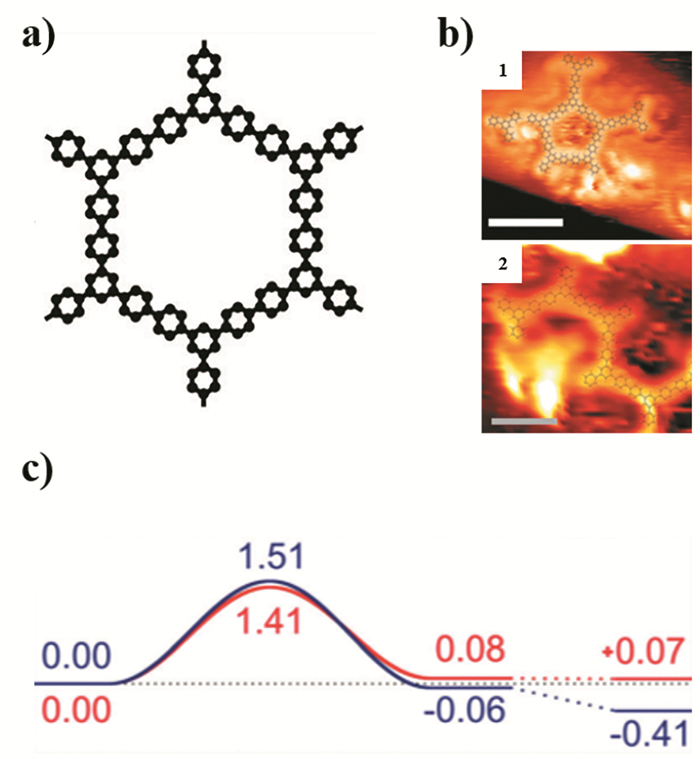
 下载:
下载:
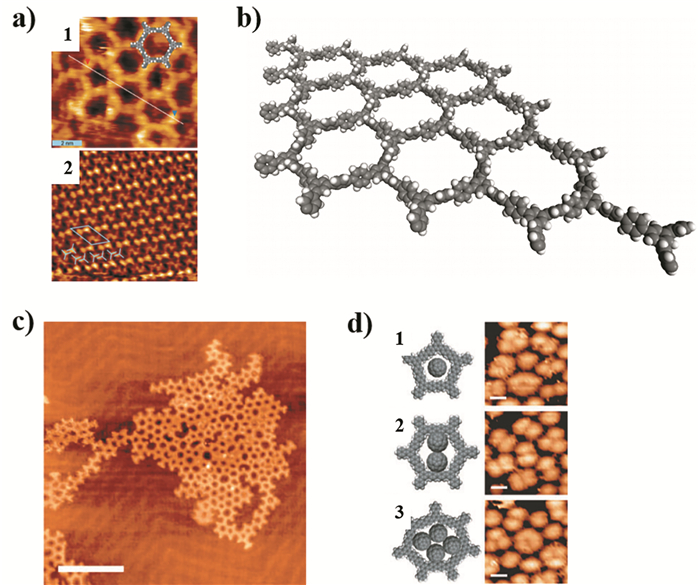
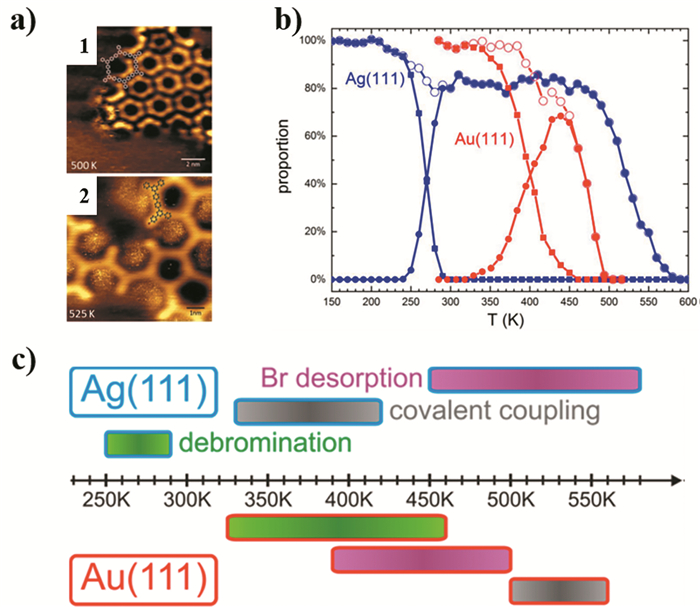




 下载:
下载:















