引用本文:
郭天雨, 刘粟侥, 青明, 冯景丽, 吕振刚, 王洪, 杨勇. 原位XRD反应装置下H2O对Fe5C2的物相及F-T反应性能影响的研究[J]. 燃料化学学报,
2020, 48(1): 75-82.
Citation:
GUO Tian-yu, LIU Su-yao, QING Ming, FENG Jing-li, Lü Zhen-gang, WANG Hong, YANG Yong. In situ XRD study of the effect of H2O on Fe5C2 phase and Fischer-Tropsch performance[J]. Journal of Fuel Chemistry and Technology,
2020, 48(1): 75-82.
State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
2.
University of Chinese Academy of Sciences, Beijing 100049, China
3.
National Energy Center for Coal to Liquids, Synfuels China Co. Ltd, Beijing 101407, China
Corresponding author:
YANG Yong, Tel: +86 69667699, E-mail: yyong@sxicc.ac.cn
Received Date:
27 September 2019 Revised Date:
22 October 2019 Available Online:
01 January 2020
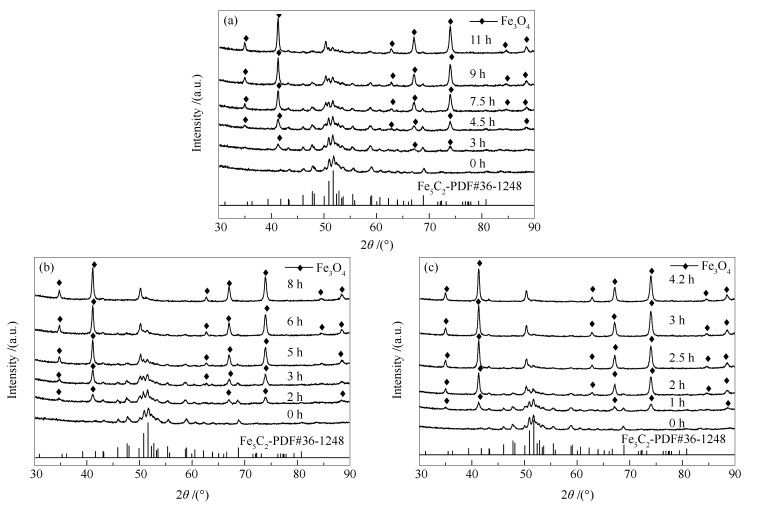
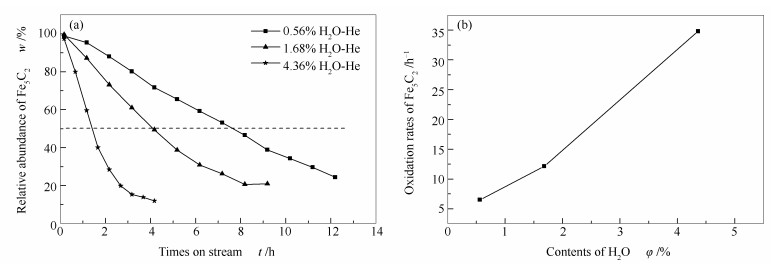
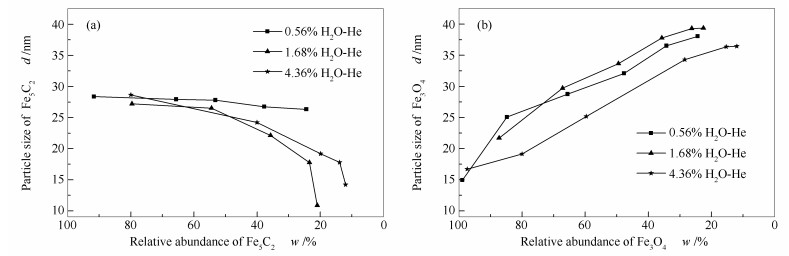
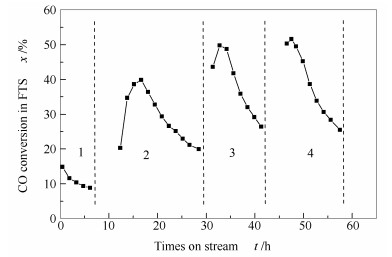
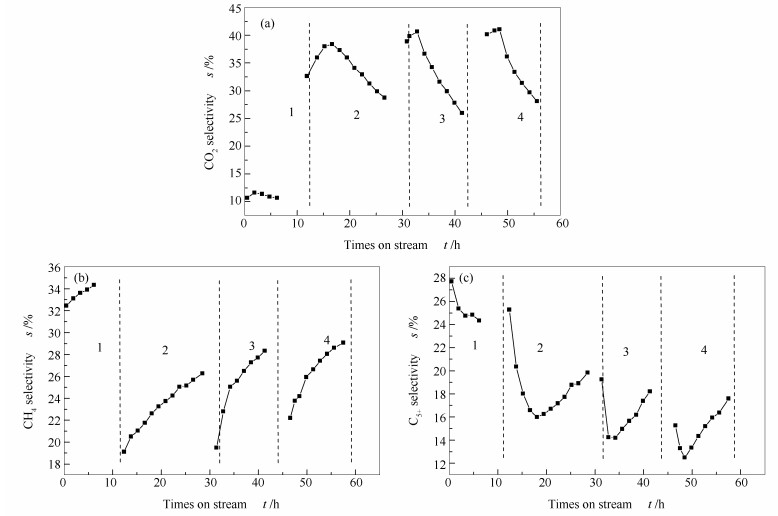
Abstract:In situ XRD reaction device combined with the online gas chromatography was used to study the oxidation behavior of the effect of H2O content (4.36%, 1.68%, 0.56%) on the phase and Fischer-Tropsch synthesis (FTS) performance of the single phase Fe5C2. The results show that the oxidation rate of the Fe5C2 phase increases with the increase of the content of injected H2O. Meanwhile, the particle size of Fe5C2 phase decreases and more active sites exposes during the H2O oxidation, resulting in the increase of the FTS activity. Furthermore, the FTS activity increases with the increase of the oxidation times, but the selectivity of CH4 increases and the C5+ selectivity decreases gradually.
RÖPER M. Fischer-Tropsch Synthesis[C]// Catalysis in C 1 Chemistry. 1983.
[3]
ANDERSO R B, KOLBE H, RALEK M. The Fischer-Tropsch Synthesis[M]. NewYork: Academic Press, 1984.
[4]
温晓东, 杨勇, 相宏伟, 焦海军, 李永旺. 费托合成铁基催化剂的设计基础:从理论走向实践[J]. 中国科学:化学,
2017,47,(11): 1298-1311.
WEN Xiao-dong, YANG Yong, XIANG Hong-wei, JIAO Hai-jun, LI Yong-wang. Design basis of fischer-tropsch synthesis of iron-based catalysts: from theory to practice[J]. Chin Sci: Chem,
2017, 47(11):
1298-1311.
[5]
WANG Y, KANG J, ZHANG Q. Research advances in catalysts for fischer-tropsch synthesis[J]. Pet Technol,
2009, 38(12):
1255-1263.
[6]
SMIT E D, WECKHUYSEN B M. ChemInform abstract: The renaissance of iron-based Fischer-Tropsch synthesis: The multifaceted catalyst deactivation behavior[J]. ChemInform,
2010, 40(19):
2758-2781.
[7]
LI S, ROBERT J O, MEITZNER G D. Structural analysis of unpromoted Fe-based Fischer-Tropsch catalysts using X-ray absorption spectroscopy[J]. Appl Catal A: Gen,
2001, 219(1):
215-222.
[8]
DUVENHAGE D J, ESPINOZA R L, COVILLE N J. fischer-tropsch precipitated iron catalysts: Deactivation studies[J]. Stud Surf Sci Catal,
1994, 88:
351-358.
doi: 10.1016/S0167-2991(08)62760-3
[9]
BUKUR D B, OKABE K, ROSYNEK M P. Activation studies with a precipitated iron catalyst for fischer-tropsch synthesis. Ⅰ: Characterization studies[J]. J Catal,
1995, 155(2):
353-365.
doi: 10.1006/jcat.1995.1217
[10]
BARTHOLOMEW C H, STOKER M W, MANSKER L. Effects of pretreatment, reaction, and promoter on microphase structure and fischer-tropsch activity of precipitated iron catalysts[J]. Stud Surf Sci Catal,
1999, 126:
265-272.
doi: 10.1016/S0167-2991(99)80475-3
[11]
REYMOND J P, MERIAUDEAU P, TEICHNER S J. Changes in the surface structure and composition of an iron catalyst of reduced or unreduced Fe2O3 during the reaction of carbon monoxide and hydrogen[J]. J Catal,
1982, 75(1):
39-48.
[12]
BUTT J B. Carbide phases on iron-based fischer-tropsch synthesis catalysts part Ⅰ: Characterization studies[J]. Catal Lett,
1990, 7(1/4):
61-81.
[13]
RAUPP G B, DELGASS W N. Mössbauer investigation of supported Fe and FeNi catalysts: Ⅱ. Carbides formed fischer-tropsch synthesis[J]. J Catal,
1979, 58(3):
348-360.
doi: 10.1016/0021-9517(79)90274-4
[14]
MACHOCKI K. Formation of carbonaceous deposit and its effect on carbon monoxide hydrogenation on iron-based catalysts[J]. Appl Catal: Gen,
1991, 70(1):
237-252.
doi: 10.1016/S0166-9834(00)84167-6
[15]
DWYER D J, HARDENBERGH J H. The catalytic reduction of carbon monoxide over iron surfaces: A surface science investigation[J]. Chem Inform,
1984, 87(1):
66-76.
[16]
DRY M E. Catalysis-Science and Technology[M]. NewYork: Springer Verlag, 1988: 160-255.
[17]
MANSKER L D, JIN Y, BUKUR D B. Characterization of slurry phase iron catalysts for fischer-tropsch synthesis[J]. Appl Catal A: Gen,
1999, 186(s 1/2):
277-296.
[18]
WELLER S, HOFER L J E, ANDERSON R B. The role of bulk cobalt carbide in the Fischer-Tropsch synthesis1[J]. J Am Chem Soc,
1948, (2):
799-801.
[19]
BUKUR D B, NOWICKI L, MANNE R K. Activation studies with a precipitated iron catalyst for Fischer-Tropsch synthesis: Ⅱ. Reaction studies[J]. J Catal,
1995, 155(2):
366-375.
doi: 10.1006/jcat.1995.1218
[20]
BARTHOLOMEW C H, BOWMAN R M. Sulfur poisoning of cobalt and iron Fischer-Tropsch catalysts[J]. Appl Catal A: Gen,
1985, 15(1):
59-67.
doi: 10.1016/S0166-9834(00)81487-6
[21]
KRITZINGER J A. The role of sulfur in commercial iron-based Fischer-Tropsch catalysis with focus on C2-product selectivity and yield[J]. Catal Today,
2002, 71(3):
307-318.
[22]
PENDYALA V R R, JACOBS G, MOHANDAS J C. Fischer-Tropsch synthesis: Effect of water over iron-based catalysts[J]. Catal Lett,
2010, 140(3/4):
98-105.
[23]
ANDERSON R.B. Kinetics of the Fischer-Tropsch synthesis on iron catalysts[J]. Synfacts,
1964, 44(2):
1065-1070.
[24]
SATTERFIELD C N, HANLON R T, TUNG S E. Effect of water on the iron-catalyzed Fischer-Tropsch synthesis[J]. Ind Eng Chem Prod Res Dev,
1986, 25(3):
407-414.
doi: 10.1021/i300023a007
[25]
BELL W K, HAAG W O. Conversion of synthesis gas to liquid hydrocarbons gel: US 4978689[P]. 1990-12-18.
[26]
GALVISl H M T, BITTER J H, DAVIDIAN T. Iron particle size effects for direct production of lower olefins from synthesis gas[J]. J Am Chem Soc,
2012, 134(39):
16207-16215.
doi: 10.1021/ja304958u
[27]
ZHANG H B, SCHRADER G L. Characterization of a fused iron catalyst for Fischer-Tropsch synthesis by in situ laser Raman spectroscopy[J]. J Catal,
1985, 95(1):
325-332.
doi: 10.1016/0021-9517(85)90038-7
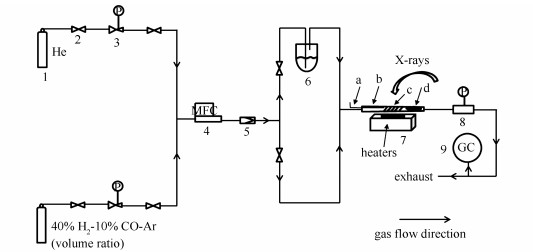
图 1
原位XRD反应装置示意图
Figure 1
Schematic diagram of the in situ XRD reactor
1: normal gas cylinder; 2: needle valve; 3: pressure reducing valve; 4: mass flow controller; 5: check valve; 6: bubbler; 7: in situ capillary reactor setup (a: thermocouple, b: silica capillary, c: catalyst sample, d: silica wool); 8: back pressure valve; 9: gas chromatography (agilent 6890 N)

 下载:
下载:

 下载:
下载:















