School of Biomedical Engineering, Dalian University of Technology, Dalian 116024, China
b.
CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
c.
College of Chemistry and Chemical Engineering, Wuhan Textile University, Wuhan 430200, China
* Corresponding author at: Dalian Institute of Chemical Physics
Chinese Academy of Sciences
Dalian 116023
China. E-mail address: qinggy@dicp.ac.cn (G. Qing). 1 These authors contributed equally to this work.
Received Date:
25 October 2021 Accepted Date:
14 March 2022 Revised Date:
31 January 2022 Available Online:
15 February 2023
Abstract:
Prion diseases are fatal neurodegenerative diseases that can cause severe dementia. The misfolding and accumulation of the prion peptide (PrP)106–126 is crucial, and this process is closely relevant to biological membranes. However, how PrP106–126 aggregation is affected by the molecular chirality of phospholipid membrane is unknown. Thus, in this study, a pair of L- and D-aspartic acid (Asp)-modified 1, 2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) were synthesized to construct chiral liposomes. We discover that L-Asp-DPPE liposomes strongly inhibit the oligomerization and amyloidogenesis of PrP106–126, whether acting on monomers or oligomers, which rescues cytotoxicity induced by PrP106–126. By comparison, D-Asp-DPPE liposomes inhibit peptide oligomerization only at a high concentration and cannot prevent amyloidogenesis when acting on oligomers, which lead to pronounced cytotoxicity. Apoptosis experiment, dynamic change of intracellular Ca2+ (iCa2+) and Ca2+ release from endoplasmic reticulum (ER), reactive oxygen species (ROS) production, adsorption dynamics and affinity tests, and fluorescent imaging clearly disclose that molecular chirality of the liposomes dominates conformational transition of PrP106–126 from random coil to β-sheet, binding and adsorption of the monomers and oligomers, and subsequent fibrillation process, resulting in distinct inhibition effect in Ca2+ overload and release, ROS production and cell apoptosis. This work is the first to report that interfacial molecular chirality is a potentially crucial influence on the fibrillation process of PrP106–126 and its cell responses, whereas the convergence of chiral amino acids and liposomes can be considered potential inhibitors in prion diseases.
Prions are infamous proteins that are devoid of nucleic acids that can cause infectious and rapidly progressive neurodegenerative diseases, such as Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker syndrome and fatal familial insomnia in humans; bovine spongiform encephalopathy in cattle; and chronic wasting disease in deer and elk [1]. Despite the relatively low transmission frequency of prion diseases, they have been the subject of much attention due to their long silent incubation period and high fatality rate, which pose an unpredictable risk to disease control and public health [2]. Importantly, scholars have intensively studied prion diseases because prion-like mechanisms might also apply to many other neurodegenerative diseases, such as tauopathies, Huntington disease, Alzheimer's disease, Parkinson disease, and amyotrophic lateral sclerosis [3-6].
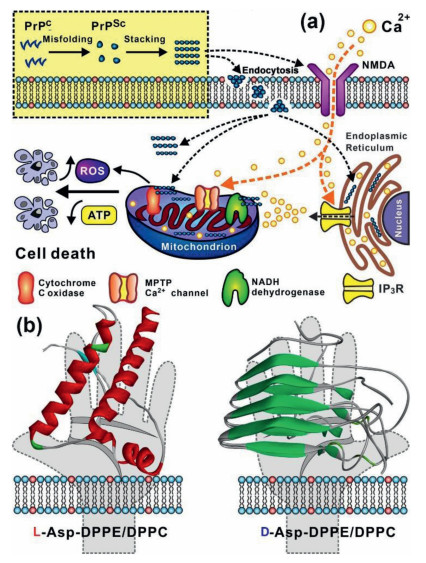
The main feature of prion diseases is the accumulation of PrPSc (scrapie isoform of the prion protein) in the brain, which has more β-sheet content relative to the normal cellular form of PrPC. PrPSc exhibits strong aggregation propensities and contributes to neurotoxicity [1, 7, 8]. PrPSc neurotoxicity has been reported to be closely associated with Ca2+ dyshomeostasis [9, 10] and mitochondrial malfunction (Fig. 1a) [11, 12]. Among the many forms of prion peptides, PrP106–126 (KTNMKHMAGAAAAGAVVGGLG) is recognized as a main domain involved in conformation conversion and as being highly aggregated into amyloid fibers, generating neurotoxicity both in vivo [13] and in vitro [14, 15]. Therefore, approaches that inhibit the aggregation of PrP106–126 would be responsible for the treatment of prion diseases.
Figure 1
Figure 1.
Remarkable influence of phospholipid membrane on prion fibrillation and its damage to mitochondria. (a) PrPC misfolds to PrPSc on the cell membrane and aggregates into oligomers and long fibrils. On the one hand, PrPSc can affect the N-methyl-D-aspartate (NMDA) receptor channel, which is permeable to Ca2+; on the other hand, PrPSc can be internalized by the cell membrane and accumulate in the endoplasmic reticulum (ER), leading to perturbation in Ca2+ homeostasis. Ca2+ accumulation into the mitochondria results in ROS production, decreased of ATP production and the release of cytochrome c, all of which are closely associated with cell apoptosis and death. (b) Graphic illustration of effect of molecular chirality of L- and D-Asp-DPPE/DPPC liposomes on PrP misfolding.
The pathophysiology of prion diseases appears to lie in a misfolding process from caveolae-like domains [16]. Because caveolae are enriched in lipids such as sphingolipids, it is highly necessary to study the interaction of PrP106–126 with biological membranes, especially the phospholipid membrane in prion diseases. Studies have shown that PrPC binds to membranes that contain negatively charge to become β-sheet rich structures [17-19]. However, how the chirality of phospholipid membrane affects prion diseases has been rarely investigated. In general, natural phospholipid molecules exhibit a strong preference for their L-enantiomers, and the cell membranes are covered with numerous chiral biomolecules, such as amino acids, peptides, proteins, and polysaccharides. These chiral biomolecules are either embedded into the phospholipid bilayers or modified on the tip of the phospholipid molecules. Importantly, amino acid chirality has been recognized as a main driving force in determining the orientation of side chains, folding of the peptide backbone, specific binding with guests through stereoselective hydrogen bonding interactions, and even the bio-function of proteins [20, 21]. Therefore, examining the effect of chiral amino acid modified phospholipids on the misfolding and aggregation of PrP106–126 is crucial (Fig. 1b).
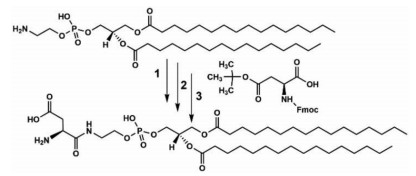
In this study, chiral aspartic acid (Asp) was selected to modify DPPE phospholipid because Asp-plays crucial roles in neuroendocrine systems, meanwhile Asp-is an amino acid with two carboxylic acid group, the β-carboxylic acid was attached to the head amine of DPPE through condensation reaction, which remained a pair of carboxylic acid and amine capable of guaranteeing amphiphilicity of the Asp-modified phospholipid. Specifically, a pair of L- and D-Asp‒modified 1, 2-dipalmitoyl-sn-glycero-3-phosphoethanol-amine (abbreviated to L/D-Asp-DPPE, Scheme 1) with satisfactory mirror symmetry were synthesized in our laboratory [22]. L/D-Asp-DPPE was first used to construct liposomes to investigate the effect of liposomes chirality on the PrP106–126 aggregation. L- or D-Asp-DPPE was allowed to mix with an equivalent amount of 1, 2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) to construct liposomes with a uniform particle size of 100 nm through a classical extrusion method. When the self-assembled L- or D-Asp-DPPE/DPPC liposomes were incubated with PrP106–126, remarkable chiral discrimination was detected in inhibiting PrP106–126 oligomerization and fibrillation, which then led to a clear difference in Ca2+ homeostasis and ROS production in mouse neuroblastoma (N2a) cells (a typical cell line for neurodegenerative diseases). The detailed mechanism underlying how aggregation was inhibited was analyzed using adsorption dynamics, affinity evaluation and fluorescence co-localization, indicating that the binding capacity of PrP106–126 on the L-Asp-DPPE phospholipid surface was substantially higher than that on the D-surface whether within or without the cellular milieu, which caused L-Asp-DPPE liposomes to perform better than D-Asp-DPPE liposomes in inhibiting PrP106–126 fibrillation and in rescuing fibrillation-induced cytotoxicity.
Scheme 1
Scheme 1.
Synthetic route of L-Asp-DPPE, reaction conditions: (1) EDC/HOBT/DMAP, solvent: chloroform, 50 ℃, 24 h; (2) piperidine and chloroform (v/v = 1:4), r.t., 1 h; (3) TFA/Tis/H2O (v/v/v = 95:2.5:2.5), r.t., 1 h. D-Asp-DPPE was synthesized according to the same method except Fmoc-D-Asp(OtBu)-OH was used.
This study provides a novel insight into the crucial fibrillation process of prion protein from the perspective of molecular chirality. Specifically, we report that chiral liposomes exhibit promise and excellent biocompatibility in the treatment of prion diseases. Importantly, we propose a reasonable explanation for the differences in cellular responses to prion peptide and chiral liposomes by dynamically detecting of both iCa2+ and ER-Ca2+. This bridges the large gap between the properties of material interface and cellular behaviors, thus aiding the development of therapeutic strategies for neurodegenerative diseases.
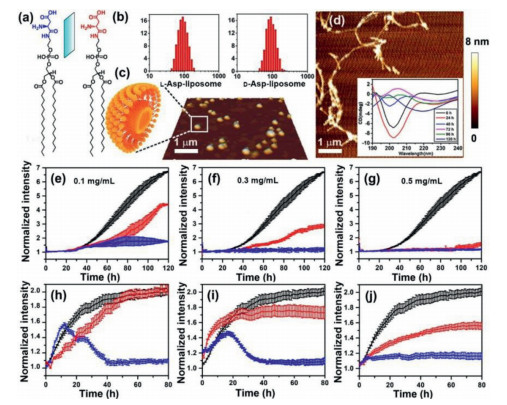
L- and D-Asp-DPPE with satisfactory mirror symmetry were shown in Fig. 2a. A lipid extrusion apparatus [23] (Fig. S1a in Supporting information) was used to obtain L- or D-Asp-DPPE/DPPC liposomes (abbreviated as L- or D-Asp-liposomes unless otherwise indicated) with a narrow size distribution of approximately 100 nm, which was confirmed by dynamic light scattering (Fig. 2b) and atomic force microscopy (AFM, Fig. 2c). Notably, DPPC was introduced for mixing with L- or D-Asp-DPPE to improve the stability of the liposomes. Furthermore, the CD spectra revealed that L- and D-Asp-liposomes had favorable mirror symmetry and their chiralities were mainly derived from L- or D-Asp (Fig. S1b in Supporting information).
Figure 2
Figure 2.
Characterization of liposomes, fibrils of PrP106–126, and influence of molecular chirality of L- or D-Asp-DPPE liposomes on fibrillation of PrP106–126 monomers and oligomers. (a) Chemical structures of L- and D-Asp-DPPE. (b) Size distribution of L- and D-Asp-liposomes analyzed by dynamic laser scattering. (c) Three-dimensional AFM image of prepared liposomes. (d) AFM image obtained after incubation of PrP106–126 monomers at 37 ℃ for 120 h. Inset shows time dependence of CD spectra of PrP106–126, verifying the conformational transition of the peptide from a random coil to β-sheet state. (e–j) ThT-monitoring kinetic curves of PrP106–126 fibrillation at 37 ℃. Black lines: PrP106–126 (e–g) monomers or (h–j) oligomers alone; blue or red lines: PrP106–126 monomers or oligomers incubated with L- (blue) or D- (red) Asp-liposomes. PrP106–126 concentrations were 100 µmol/L, and the L- or D-Asp-liposomes concentrations were (e, h) 0.1, (f, i) 0.3 and (g, j) 0.5 mg/mL, respectively. The test solutions were 150 mmol/L NaCl. The error bars represent standard deviations from the mean of at least three independent experiments.
On the other hand, after 120 h incubation at 37 ℃, PrP106–126 (Synpeptide Co., Ltd; final concentration was 100 mmol/L in 150 mmol/L NaCl) could aggregate into many long β-sheet-rich fibrils, as observed by AFM (Fig. 2d). The CD spectra indicated the conformational transition of PrP106–126 during this long fibrillation process [24], where PrP106–126 changed from being in a random coil state to a having β-sheet structure (Fig. 2d inset). The fibrillation process of PrP106–126 could be divided into two phases, namely a nucleation phase in which monomers misfold and aggregate to oligomers and an elongation phase to allow the growth of the oligomers to long fibers [25]. The oligomers of the peptide are suggested to be the major toxic form involved in protein conformational diseases and have attracted increasing attention in recent years [26, 27].
Therefore, the effects of L- or D-Asp-liposomes on the fibrillation processes of the monomers and oligomers of PrP106–126, respectively, were investigated. First, the influence of L- or D-Asp-liposomes addition in the nucleation phase of PrP106–126 was evaluated, the fibrillation kinetics were monitored using a standard thioflavine-T (ThT) binding fluorescence assay [28]. In this experiment, L- or D-Asp-liposomes with different concentrations (final concentration was 0.1, 0.3, or 0.5 mg/mL) were added to the PrP106–126 monomers solution (final concentration was 100 µmol/L in 150 mmol/L NaCl) at the beginning of incubation at 37 ℃, respectively. According to the dynamic growth curve, as shown by black lines in Figs. 2e-g, PrP106–126 began to aggregate at 30 h and completed fibrillation after 120 h. As for the addition of L-Asp-liposomes, the aggregation of PrP106–126 was remarkably suppressed (Figs. 2e-g, blue lines), even though a low concentration of L-Asp-liposomes (0.1 mg/mL) was used. By contrast, only the high concentration of D-Asp-liposomes (0.5 mg/mL) could suppress the PrP106–126 fibrillation, where a decrease in D-Asp-liposomes dosage contributed the most to reducing the inhibitory effect.
Subsequently, we investigated the influence of L- or D-Asp-liposomes on the fibrillation of PrP106–126 oligomers. Before this experiment, PrP106–126 monomers were incubated at 37 ℃ for 48 h to enable the formation of PrP106–126 oligomers, which were featured with few short fibers observed by transmission electron microscope (Fig. S2 in Supporting information). As shown by the black lines in Figs. 2h-j, oligomerized PrP106–126 alone completely fiberized after 80 h. The addition of L-Asp-liposomes remarkably inhibited the aggregation of PrP106–126 oligomers and displayed a dosage-dependent inhibitory effect. A high concentration of L-Asp-liposomes (0.5 mg/mL) completely suppressed the growth of PrP106–126 oligomers. However, the inhibitory effects were substantially weaker when L-Asp-liposomes with low concentrations (0.1 and 0.3 mg/mL were used, which are indicated by clear inflection points at 18 h in the growth curves (Figs. 2h and i, blue lines). The inflection points revealed that there might be a competition between the peptide fibrillation and the inhibitory effect induced by L-Asp-liposomes with low concentrations. In sharp contrast to this, the addition of D-Asp-liposomes (0.1 and 0.3 mg/mL) did not influence the fibrillation of the PrP106–126 oligomers and only the addition of a high concentration of D-Asp-liposomes (0.5 mg/mL) could slightly inhibit this process (Fig. 2j, red line). These results clearly indicated that L-Asp-liposomes remarkably prevented PrP106–126 fibrillation by acting on either monomers or oligomers. Conversely, D-Asp-liposomes slightly inhibited the fibrillation of PrP106–126 monomers. The inhibitory effect depended on the liposome concentrations, and D-Asp-liposomes barely influenced the fibrillation of PrP106–126 oligomers.
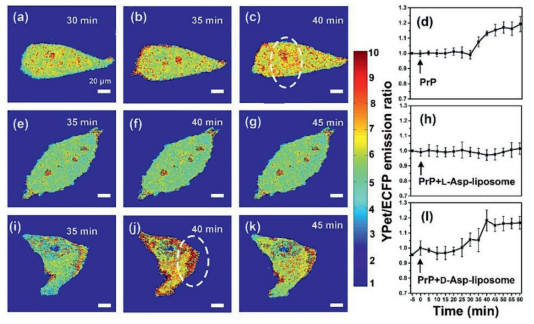
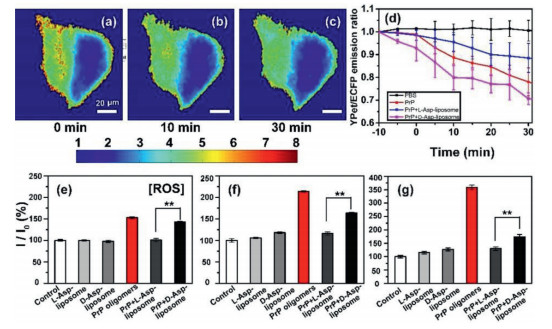
Frist, the effect of L- or D-Asp-liposomes on PrP106–126-induced cytotoxicity and apoptosis was investigated by using N2a cells. As shown in Fig. S3 (Supporting information), CCK-8 results demonstrated that both L- and D-Asp-liposomes had satisfactory biocompatibility with no toxicity to N2a cells and L-Asp-liposomes were more effective in preventing fiber formation and in reducing the cytotoxicity of PrP106–126 aggregation relative to D-Asp-liposomes. Meanwhile, flow cytometry experiment also indicated that L- and D-Asp-liposomes significantly differed in rescuing cells from the cell apoptosis caused by PrP106–126 fibrillation (Fig. S4 in Supporting information). Ca2+ is closely related to the process of neurochemical signaling [29]. PrP106–126 are involved in the neuronal loss that occurs in prion diseases, partially due to calcium dyshomeostasis [30], and calcium dyshomeostasis may be an early symptom of most neurodegenerative diseases. To investigate why a chiral difference was present in the rescue of PrP106–126-induced apoptosis, we monitored the concentration of cytoplasmic Ca2+ produced. To do so, we used a high sensitivity fluorescence resonance energy transfer (FRET)-based Cyto-Ca2+ biosensor to measure the cytoplasmic Ca2+ at the level of a single cell. The construction of the FRET-based Ca2+ biosensor (i.e., ECFP-CaM-M13-EYFP) was described in a previous study [31]. Differently, EYFP was replaced by a recently developed YFP variant YPet [32]. Fig. S5 (Supporting information) illustrates the activation mechanism of the FRET Cyto-Ca2+ biosensor. The fluorescent intensity ratio of YPet to ECFP represents the concentration of Ca2+ production in cells; the calculation method is detailed in Supplementary Part. Initially, the FRET Cyto-Ca2+ biosensor was transfected into the N2a cells by using transfection reagent, per the manufacturer's instructions. The N2a cells treated with PBS were used as a control, in which no evident change in the concentration of iCa2+ was detected (Fig. S6 in Supporting information). Subsequently, we added 100 µmol/L of PrP106–126 oligomers to the cell culture medium to study its effect on Ca2+ homeostasis. As shown in Figs. 3a-c, after experiencing a steady phase for 30 min, the iCa2+ level began to increase and sustained for 30 min; this was indicated by a greater quantity of red dots appearing in the cell images (Figs. 3b and c), reflecting an increase in the YPet/ECFP ratio from 1.0 to 1.2 (Fig. 3d). This result indicated that PrP106–126 oligomers could cause iCa2+ overload.
Figure 3
Figure 3.
Cytoplasmic Ca2+ monitor. Fluorescence images of the distribution of cytoplasmic Ca2+ in a single N2a cell. In the first row, images were collected at (a) 30, (b) 35 and (c) 40 min after PrP106–126 oligomers (100 µmol/L) were added to the cells. In the second and third rows, images were collected at (e, i) 35, (f, j) 40 and (g, k) 45 min after a mixture of PrP106–126 with (e-g) L- or (i-k) D-Asp-liposomes was added to the cells. Color bar represents the YPet to ECFP ratio, with cool and warm colors indicating low and high concentrations of iCa2+, respectively. In the rightmost column, time-dependent YPet to ECFP ratios to show the change of the iCa2+ concentration in response to the addition of (d) PrP106–126 oligomers, mixture of PrP106–126 with (h) L- or (l) D-Asp-liposomes. Error bars represent standard deviations from the mean of at least three independent experiments. Scale bar: 20 µm.
Subsequently, L- or D-Asp-liposomes (0.3 mg/mL) were co-incubated with PrP106–126 for 48 h, and we then added the mixture to the N2a cells to study its effect on the calcium homeostasis. When the mixture of L-Asp-liposomes with PrP106–126 was added, the Cyto-Ca2+ biosensor detected no excess iCa2+ (Figs. 3e-g). The fluorescent ratio of YPet/ECFP (Fig. 3h) exhibited the same trend as the control sample treated by PBS at the same time. By contrast, when the mixture of D-Asp-liposomes with PrP106–126 was added, the concentration of iCa2+ increased at 35 min as indicated by the numerous red dots in the cell image (Figs. 3i-k). The fluorescent ratio of YPet/ECFP increased from 35 min, peaked (at 1.18) at 40 min, and then sustained for 20 min (Fig. 3l). These results clearly demonstrated that L- and D-Asp-liposomes exhibit remarkable difference in rescuing PrP106–126-induced calcium dyshomeostasis.
The accumulation of PrPSc in ER can lead to perturbations in the calcium homeostasis of the ER and then lead to iCa2+ overload. The chiral difference was further investigated in the rescue of PrP106–126 aggregation-induced perturbations of ER-Ca2+. In this study, N2a cells that were transfected with an ER-Ca2+ FRET biosensor [33] were pretreated with PrP106–126 oligomers, mixture of L- or D-Asp-liposomes with PrP106–126 (incubated at 37 ℃ for 48 h extracellular) for 30 min. As shown in Figs. 4a-c, the oligomers of PrP106–126 caused an obvious release of Ca2+ from the ER, reflecting as a gradual decrease of the fluorescent ratio of YPet/ECFP from 1.0 to 0.75 (Fig. 4d, red line). When L-Asp-liposomes and PrP106–126 were added to the N2a cells, the fluorescent ratio of YPet/ECFP only decreased to 0.87, (Fig. 4d, blue line). By contrast, this ratio decreased to 0.69 upon the addition of the mixture of D-Asp-liposomes with PrP106–126 (Fig. 4d, pink line). This result further evinced the chiral discrimination between L- and D-Asp-liposomes in terms of the ER-Ca2+.
Figure 4
Figure 4.
Chiral discrimination monitored by ER-Ca2+ and ROS production. MATLAB-processed images displaying the ER-Ca2+ concentration in a single N2a cell with additions of PrP106–126 oligomers (100 µmol/L). Images were collected at (a) 0, (b) 10 and (c) 30 min, respectively. (d) Time-dependent YPet to ECFP ratio showing the change of ER-Ca2+ concentration in response to additions of PBS (blank), PrP106–126 oligomers (red), and a mixture of PrP106–126 with L- (blue) or D-Asp-liposomes (pink). Comparison of intracellular ROS changes of N2a cells incubated with L- or D-Asp-liposomes (0.3 mg/mL), PrP106–126 oligomers (100 µmol/L), or with both PrP106–126 and L- or D-Asp-liposomes for (e) 1.5 and (f) 8 h at 37 ℃. (g) Comparison of intracellular ROS changes in N2a cells coincubated with L- or D-Asp-liposomes (0.3 mg/mL), PrP106–126 monomers (100 µmol/L), or with both PrP106–126 monomers and L- or D-Asp-liposomes for 48 h at 37 ℃. For solution-based assays, the error bars represent standard deviations from the mean of at least three independent experiments. *P < 0.05, **P < 0.01, indicating a significant difference. Scale bar: 20 µm.
After iCa2+ level increases, mitochondria can rapidly take up iCa2+ to prevent Ca2+ overload into the cytosol. However, excessive Ca2+ taken up by mitochondria can lead to mitochondrial malfunction, such an excessive reactive oxygen species (ROS) generation, which plays a central role in the pathophysiology of neurodegenerative diseases (Fig. 1a) [12, 34]. To monitor ROS production in N2a cells, we conducted a dichlorofluorescein diacetate (DCFH-DA) assay [35]. First, the liposomes (0.3 mg/mL) were incubated at 37 ℃ for 48 h before being added to the N2a cells. L- and D-Asp-liposomes did not significantly differ in the production of cellular ROS after 1.5 h (Fig. 4e) and 8 h (Fig. 4f) application, which was comparable with the ROS results for cells treated with PBS. In sharp contrast to this, when 100 µmol/L of PrP106–126 oligomers were added to the N2a cells, sharp increases of ROS production were observed after 1.5 h (53% increase, Fig. 4e) and 8 h (114% increase, Fig. 4f). The extracellular incubation of PrP106–126 with L-Asp-liposomes at 37 ℃ for 48 h could remarkably reduce ROS production after being added to the cells for 1.5 h (Fig. 4e) and 8 h (Fig. 4f), all of which maintained at the normal level comparable to the N2a cells treated with PBS alone. By comparison, the inhibitory effect of D-Asp-liposomes on PrP106–126-induced ROS production (Figs. 4e and f, black columns) was substantially weaker under the same condition. The chiral difference of liposomes in PrP106–126-induced ROS production was also obvious when the mixtures were added to the cells at the beginning of incubation and then co-cultured at 37 ℃ for 48 h (Fig. 4g). These results demonstrated the significant difference between L- and D-Asp-liposomes in suppressing PrP106–126-induced ROS production, which further indicated a significant discrepancy in cell apoptosis and cytotoxicity.
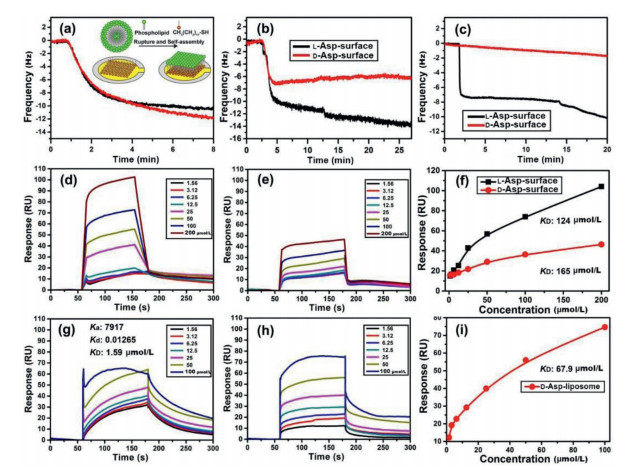
To provide direct evidence for the interactions between chiral liposomes and the PrP106–126 monomers or oligomers, tests on adsorption dynamics and binding affinity were performed. First, a quartz crystal microbalance (QCM) [36] was used to record the adsorption dynamics of PrP106–126 monomers or oligomers on L- or D-Asp-phospholipid monolayers, which were prepared from the burst and self-assembly of the liposomes on a hydrophobic 1-octadecy-lmercaptan monolayer immobilized on a gold surface of the QCM sensor (Fig. 5a, inset) [37]. Fig. 5a presents the dynamic self-assembly process of L- and D-Asp-liposomes on the sensor surface, and the changes in adsorption induced frequency (ΔF: 10 Hz) were nearly identical, corresponding to an adsorption quantity of 57 ng/cm2 according to the Sauerbrey equation. Subsequently, a solution of PrP106–126 monomers or oligomers (100 µmol/L in 150 mmol/L NaCl) was pumped into the system, to evaluate the adsorption capacity of the peptide on the L- or D-Asp-phospholipid surface. PrP106–126 monomers were found to have stronger adsorption on the L-Asp-phospholipid surface (ΔF: 13.80 Hz, Δm: 81.42 ng/cm2, Fig. 5b) than that on the D-surface (ΔF: 6.03 Hz, Δm: 35.6 ng/cm2). Similarly, PrP106–126 oligomers displayed higher adsorption quantities on the L-Asp-phospholipid surface (ΔF: 11.40 Hz; Δm: 67.3 ng/cm2, Fig. 5c) than that on the D-surface (ΔF: 1.93 Hz, Δm: 11.4 ng/cm2).
Figure 5
Figure 5.
Adsorption dynamics and binding affinity tests of PrP106–126 monomers or oligomers with L- or D-Asp-liposomes. (a) Dynamic adsorption curves of L- (black) and D-Asp-liposomes (red, 1 mg/mL) ruptured and self-assembled on the thiolated gold surface of QCM sensors (Inset of a). Dynamic curves of PrP106–126 (b) monomers or (c) oligomers adsorbed on the L- (black) or D-Asp-phospholipid (red) self-assembled surface, obtained from the QCM tests. Concentrations of PrP106–126 are 100 µmol/L in 150 mmol/L NaCl. Sensorgrams of PrP106–126 (d, e) monomers or (g, h) oligomers binding to (d, g) L- or (e, h) D-Asp-phospholipid self-assembled surface. Concentrations are 1.56, 3.12, 6.25, 12.5, 25, 50, 100 and 200 µmol/L for PrP106–126 monomers and 1.56, 3.12, 6.25, 12.5, 25, 50 and 100 µmol/L for PrP106–126 oligomers. PrP106–126 concentration dependent SPR responses and fitted curves for calculating KD for the binding of peptide (f) monomers or (i) oligomers with the L- or D-Asp-phospholipid surface by using a steady-state method. KD of PrP106–126 oligomers binding to the L-Asp-phospholipid surface was obtained using the dynamics analysis method, which is not shown in directly given by the Biacore analysis software (i).
Furthermore, the binding affinity between the PrP106–126 monomers or oligomers and the L- or D-Asp-liposomes was measured using surface plasmon resonance (SPR) [38]. Phospholipid monolayer modification of the SPR sensor was conducted per the same method as that for the QCM adsorption test. A series of PrP106–126 monomer solutions with different concentrations were successively pumped into the SPR chip that was modified with the L- or D-Asp-liposomes, and the response units were recorded (Figs. 5d and e). We used a steady-state method [39, 40] to analyze the relationship between SPR responses and the concentration of PrP106–126 monomers, where a smaller equilibrium dissociation constant (KD) corresponded to a stronger binding affinity between the peptide and liposomes. The fitting results revealed that the KD of the PrP106–126 monomers with the L-Asp-phospholipid surface was 124 µmol/L, which was smaller than that with the D-Asp-phospholipid surface (KD: 165 µmol/L; Fig. 5f). Control experiment indicated that the PrP106–126 adsorption–induced SPR response change was negligible on DPPE/DPPC phospholipid surface (Fig. S7 in Supporting information), corresponding to a weak affinity between them. Shown in Fig. 5g, for the PrP106–126 oligomers with the L-Asp-phospholipid surface, the KD (1.59 µmol/L) was obtained by using a dynamic analysis method, due to the observed dissociation curves drifted downward slowly with time, which represents a stronger combination pattern compared with that in the steady-state method [40]. With regard to the binding of PrP106–126 oligomers with the D-Asp-phospholipid surface, a KD of 67.9 µmol/L was obtained by the steady-state method (Figs. 5h and i). The substantially lower KD value suggested that the PrP106–126 oligomers had a higher binding affinity with the L-Asp-liposomes than that with the D-Asp-liposomes.
Microscale thermophoresis (MST) [41] was also used to measure the binding affinity between PrP106–126 and L- or D-Asp-liposomes in situ. Different from the QCM and SPR tests, which require the construction of the phospholipid monolayers, MST can provide direct affinity data in solution and is particularly suitable for macromolecular system like liposomes. The MST time traces of rhodamine B‒labeled PrP106–126 toward different concentrations of L- or D-Asp-liposomes were shown in Fig. S8 in Supporting information, which also revealed that PrP106–126 monomers had a stronger binding affinity with the L-Asp-liposomes (KD: 135 µmol/L), than that with the D-Asp-liposomes (KD: 298 µmol/L).
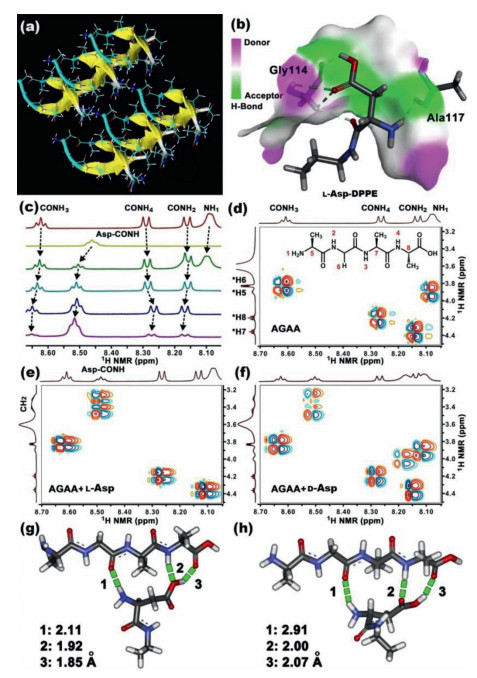
In this section, we posited a possible molecular-level explanation for chiral discrimination. Many experimental studies have shown that a hydrophobic region AGAAAAGA of PrP106–126 plays an important role in the conformational conversion from PrPC to PrPSc. Fig. 6a presents a typical packing model of the steric zipper AGAAAAGA forming amyloid fibrils [42]. Flexible molecular docking indicated that L-Asp-DPPE preferred to bind with the AGAA of PrP106–126 (Fig. 6b), which contributed to an ideal target for the molecular interaction. The binding pattern of D-Asp-DPPE with PrP106–126 is shown in Fig. S9a in Supporting information. A hydrogen nuclear magnetic resonance (1H NMR) titration experiment indicated that AGAA can form strong hydrogen bonding interactions with phenylethanamine-protected L-Asp (abbreviated to PEA L-Asp, which mimics the chiral head group of L-Asp-DPPE; Fig. S9b in Supporting information) and that most amide protons of both AGAA and PEA L-Asp-occurred clear down-field shifts as shown in Fig. 6c. By contrast, the changes in chemical shift were smaller when the complexation of AGAA and PEA D-Asp-was analyzed (Fig. S9c in Supporting information). Two dimensional 1H‒1H NMR spectra (Figs. 6d-f) further confirmed the chemical attribution of each H proton and validated the aforementioned chiral discrimination. Quantum chemistry calculations further disclosed that AGAA could form three sets of hydrogen bonds with L-Asp, and the bond lengths were substantially shorter than those of AGAA with D-Asp (Figs. 6g and h). These data indicated that the binding affinity of AGAA with L-Asp-was larger than that with its D-enantiomers. This further led to a stronger binding and adsorption of PrP106–126 on the surface of L-Asp-liposomes, resulting in the remarkable inhibition of PrP106–126 fibrillation.
Figure 6
Figure 6.
Binding mode studies. (a) Stacking model of multiple steric zipper PrP106–126 forming amyloid fibrils. Copied with permission [42]. Copyright 2011, Springer. (b) Flexible ligand docking model of L-Asp-DPPE with PrP106–126, obtained from Autodock software. The face represents the donor and acceptor of hydrogen bonding interaction for AGAAAAGA. (c) 1H NMR spectra of AGAA, PEA L-Asp, and mixtures of AGAA with 0.5, 1.0, 1.5 and 2.0 equiv. amounts of PEA L-Asp-in DMSO-d6 at 20 ℃. 1H-1H NMR spectra of AGAA (d), AGAA mixed with equimolar PEA L- (e) or D-Asp (f) in DMSO-d6 at 20 ℃. AGAA concentration was 20 mmol/L in the NMR tests. Possible interaction model of AGAA with (g) L- or (h) D-Asp, calculated from quantum chemistry calculations (Gaussian, density functional theory at 6–311 g level). Green dashed lines represent hydrogen bonds.
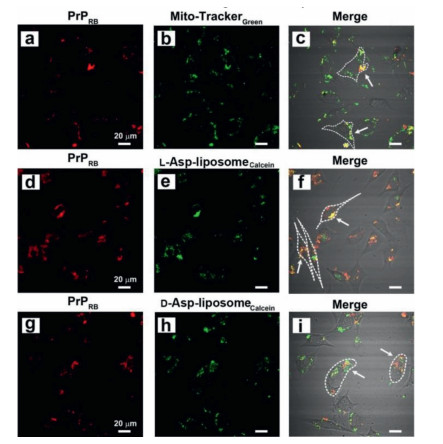
Although the aforementioned affinity tests verified the extracellular interactions between the chiral liposomes and PrP106–126, whether the interaction was maintained in the intracellular milieu was unclear. Thus, laser scanning confocal microscopy (LSCM) was used to monitor the interactions between chiral liposomes and PrP106–126 in N2a cells. L- and D-Asp-liposomes were loaded with calcein (L- or D-Asp-liposomescalcein) [43, 44] and PrP106–126 was labeled with rhodamine B (PrPRB). First, the L- or D-Asp-liposomescalein (0.3 mg/mL) were cocultured with the N2a cells for 48 h. Figs. S10a, b, d, and e in Supporting information present the distributions of the L- and D-Asp-liposomescalcein (green) and the mitochondria (red), respectively. The overlap of these LSCM figures (Figs. S10c and f in Supporting information) clearly demonstrated that L- or D-Asp-liposomes could be internalized by N2a cells, partly might fusion with the membrane of cell, and locate in the mitochondria, indicating the satisfactory biocompatibility of the chiral liposomes. By contrast, obvious cytotoxicity of PrP106–126 monomers (70 µmol/L PrP106–126 + 30 µmol/L PrP106–126RB) was observed when the peptide was incubated with the N2a cells for 48 h (Figs. 7a-c). PrP106–126 could clearly penetrate into the cells and locate in the mitochondria, as indicated by the white arrows in Fig. 7c, and remarkable mitochondria swelling occurred in most cells, which is a typical characteristic of apoptotic cells [45, 46].
Figure 7
Figure 7.
LCSM images of N2a cells incubated with PrP106–126 monomers (a-c), PrP106–126 monomers with L- (D-f) or D- (g-i) Asp-liposomes (0.3 mg/mL) for 48 h. (a, d, g) Images excited by a 559 nm laser to show the distribution of PrP106–126, (b, e, h) images excited by 488 nm laser to show the distribution of mitochondria or L- or D-Asp-liposomes, (c, f, i) merged LCSM images and optical images. Typical cell morphology is indicated by the white dashed line, whereas the overlapping regions between PrP106–126 and mitochondria or liposomes are indicated by white arrows. PrP106–126 monomers contain 70 µmol/L PrP106–126 and 30 µmol/L rhodamine B-labeled PrP106–126. The fluorescence tests were performed in triplicate for reproducibility. Scale bar: 20 µm.
When L- or D-Asp-liposomescalcein (0.3 mg/mL) and PrP106–126 monomers (70 µmol/L PrP106–126 + 30 µmol/L PrP106–126RB) were cocultured with the N2a cells for 48 h, remarkable differences in cell morphology were detected. When L-Asp-liposomes were added, most cells maintained good morphology with a long neurite (Figs. 7d-f). By comparison, when D-Asp-liposomes were added, half of the cells retracted and become round, which indicated that these cells were in the early apoptotic stage (Figs. 7g-i). Importantly, both PrP106–126 and L- or D-Asp-liposomescalcein were located in the mitochondria. Moreover, the Pearson's correlation coefficient (PCC) values described the extent of colocalization between PrP106–126 and the chiral liposomes [47]. The PCC value of L-Asp-liposomes toward PrP106–126 was 0.53, which was twice than that of D-Asp-liposomes (PCC = 0.25; Fig. S11 in Supporting information), indicating that a greater quantity of PrP106–126 was colocated with L-Asp-liposomes than that with the D-Asp-liposomes. Strong interactions between PrP106–126 and L-Asp-liposomes inhibited the peptide fibrillation, which substantially reduced the mitochondrial damage and finally rescued the cells from death.
PrP peptides are abundant in the cell membrane. The understanding of these peptides with a phospholipid membrane is crucial for elucidating the pathological progression of prion diseases. In the present study, we designed a pair of chiral amino acid–modified phospholipids, which exhibited a remarkable difference in molecular chirality when inhibiting PrP peptide aggregation and rescuing fibrillation-induced cytotoxicity. Unlike conventional studies that have focused on the physical or chemical properties of the phospholipid surface, including hydrophilicity or hydrophobicity [48] or electrical charge [49], this study revealed that the chirality of amino acid–modified phospholipids, as natural substrates for the PrP fibrillation, is a potential factor in the development of prion diseases, which provided a novel perspective to understand the crucial PrP fibrillation process from the interfacial molecular chirality [50, 51].
Importantly, phospholipids have excellent and convincible biocompatibility, and the satisfactory inhibitory effect of our chiral liposomes for the early prevention and treatment of prion diseases clearly indicate that the combination of phospholipids with chiral molecules, such as amino acids, oligopeptides and other inhibitors [52-58], might be a feasible route for developing potential drugs for the early prevention and treatment of various diseases. Furthermore, this study established a clear and direct relationship between interfacial molecular chirality and cellular behavior through the analysis of Ca2+ homeostasis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21775116 and 21922411), DICP Innovation Funding (Nos. DICP-RC201801 and DICP I202008) and LiaoNing Revitalization Talents Program (No. XLYC1802109).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.055.
Figure 1
Remarkable influence of phospholipid membrane on prion fibrillation and its damage to mitochondria. (a) PrPC misfolds to PrPSc on the cell membrane and aggregates into oligomers and long fibrils. On the one hand, PrPSc can affect the N-methyl-D-aspartate (NMDA) receptor channel, which is permeable to Ca2+; on the other hand, PrPSc can be internalized by the cell membrane and accumulate in the endoplasmic reticulum (ER), leading to perturbation in Ca2+ homeostasis. Ca2+ accumulation into the mitochondria results in ROS production, decreased of ATP production and the release of cytochrome c, all of which are closely associated with cell apoptosis and death. (b) Graphic illustration of effect of molecular chirality of L- and D-Asp-DPPE/DPPC liposomes on PrP misfolding.
Figure 2
Characterization of liposomes, fibrils of PrP106–126, and influence of molecular chirality of L- or D-Asp-DPPE liposomes on fibrillation of PrP106–126 monomers and oligomers. (a) Chemical structures of L- and D-Asp-DPPE. (b) Size distribution of L- and D-Asp-liposomes analyzed by dynamic laser scattering. (c) Three-dimensional AFM image of prepared liposomes. (d) AFM image obtained after incubation of PrP106–126 monomers at 37 ℃ for 120 h. Inset shows time dependence of CD spectra of PrP106–126, verifying the conformational transition of the peptide from a random coil to β-sheet state. (e–j) ThT-monitoring kinetic curves of PrP106–126 fibrillation at 37 ℃. Black lines: PrP106–126 (e–g) monomers or (h–j) oligomers alone; blue or red lines: PrP106–126 monomers or oligomers incubated with L- (blue) or D- (red) Asp-liposomes. PrP106–126 concentrations were 100 µmol/L, and the L- or D-Asp-liposomes concentrations were (e, h) 0.1, (f, i) 0.3 and (g, j) 0.5 mg/mL, respectively. The test solutions were 150 mmol/L NaCl. The error bars represent standard deviations from the mean of at least three independent experiments.
Figure 3
Cytoplasmic Ca2+ monitor. Fluorescence images of the distribution of cytoplasmic Ca2+ in a single N2a cell. In the first row, images were collected at (a) 30, (b) 35 and (c) 40 min after PrP106–126 oligomers (100 µmol/L) were added to the cells. In the second and third rows, images were collected at (e, i) 35, (f, j) 40 and (g, k) 45 min after a mixture of PrP106–126 with (e-g) L- or (i-k) D-Asp-liposomes was added to the cells. Color bar represents the YPet to ECFP ratio, with cool and warm colors indicating low and high concentrations of iCa2+, respectively. In the rightmost column, time-dependent YPet to ECFP ratios to show the change of the iCa2+ concentration in response to the addition of (d) PrP106–126 oligomers, mixture of PrP106–126 with (h) L- or (l) D-Asp-liposomes. Error bars represent standard deviations from the mean of at least three independent experiments. Scale bar: 20 µm.
Figure 4
Chiral discrimination monitored by ER-Ca2+ and ROS production. MATLAB-processed images displaying the ER-Ca2+ concentration in a single N2a cell with additions of PrP106–126 oligomers (100 µmol/L). Images were collected at (a) 0, (b) 10 and (c) 30 min, respectively. (d) Time-dependent YPet to ECFP ratio showing the change of ER-Ca2+ concentration in response to additions of PBS (blank), PrP106–126 oligomers (red), and a mixture of PrP106–126 with L- (blue) or D-Asp-liposomes (pink). Comparison of intracellular ROS changes of N2a cells incubated with L- or D-Asp-liposomes (0.3 mg/mL), PrP106–126 oligomers (100 µmol/L), or with both PrP106–126 and L- or D-Asp-liposomes for (e) 1.5 and (f) 8 h at 37 ℃. (g) Comparison of intracellular ROS changes in N2a cells coincubated with L- or D-Asp-liposomes (0.3 mg/mL), PrP106–126 monomers (100 µmol/L), or with both PrP106–126 monomers and L- or D-Asp-liposomes for 48 h at 37 ℃. For solution-based assays, the error bars represent standard deviations from the mean of at least three independent experiments. *P < 0.05, **P < 0.01, indicating a significant difference. Scale bar: 20 µm.
Figure 5
Adsorption dynamics and binding affinity tests of PrP106–126 monomers or oligomers with L- or D-Asp-liposomes. (a) Dynamic adsorption curves of L- (black) and D-Asp-liposomes (red, 1 mg/mL) ruptured and self-assembled on the thiolated gold surface of QCM sensors (Inset of a). Dynamic curves of PrP106–126 (b) monomers or (c) oligomers adsorbed on the L- (black) or D-Asp-phospholipid (red) self-assembled surface, obtained from the QCM tests. Concentrations of PrP106–126 are 100 µmol/L in 150 mmol/L NaCl. Sensorgrams of PrP106–126 (d, e) monomers or (g, h) oligomers binding to (d, g) L- or (e, h) D-Asp-phospholipid self-assembled surface. Concentrations are 1.56, 3.12, 6.25, 12.5, 25, 50, 100 and 200 µmol/L for PrP106–126 monomers and 1.56, 3.12, 6.25, 12.5, 25, 50 and 100 µmol/L for PrP106–126 oligomers. PrP106–126 concentration dependent SPR responses and fitted curves for calculating KD for the binding of peptide (f) monomers or (i) oligomers with the L- or D-Asp-phospholipid surface by using a steady-state method. KD of PrP106–126 oligomers binding to the L-Asp-phospholipid surface was obtained using the dynamics analysis method, which is not shown in directly given by the Biacore analysis software (i).
Figure 6
Binding mode studies. (a) Stacking model of multiple steric zipper PrP106–126 forming amyloid fibrils. Copied with permission [42]. Copyright 2011, Springer. (b) Flexible ligand docking model of L-Asp-DPPE with PrP106–126, obtained from Autodock software. The face represents the donor and acceptor of hydrogen bonding interaction for AGAAAAGA. (c) 1H NMR spectra of AGAA, PEA L-Asp, and mixtures of AGAA with 0.5, 1.0, 1.5 and 2.0 equiv. amounts of PEA L-Asp-in DMSO-d6 at 20 ℃. 1H-1H NMR spectra of AGAA (d), AGAA mixed with equimolar PEA L- (e) or D-Asp (f) in DMSO-d6 at 20 ℃. AGAA concentration was 20 mmol/L in the NMR tests. Possible interaction model of AGAA with (g) L- or (h) D-Asp, calculated from quantum chemistry calculations (Gaussian, density functional theory at 6–311 g level). Green dashed lines represent hydrogen bonds.
Figure 7
LCSM images of N2a cells incubated with PrP106–126 monomers (a-c), PrP106–126 monomers with L- (D-f) or D- (g-i) Asp-liposomes (0.3 mg/mL) for 48 h. (a, d, g) Images excited by a 559 nm laser to show the distribution of PrP106–126, (b, e, h) images excited by 488 nm laser to show the distribution of mitochondria or L- or D-Asp-liposomes, (c, f, i) merged LCSM images and optical images. Typical cell morphology is indicated by the white dashed line, whereas the overlapping regions between PrP106–126 and mitochondria or liposomes are indicated by white arrows. PrP106–126 monomers contain 70 µmol/L PrP106–126 and 30 µmol/L rhodamine B-labeled PrP106–126. The fluorescence tests were performed in triplicate for reproducibility. Scale bar: 20 µm.

 DownLoad:
DownLoad:

 下载:
下载:









