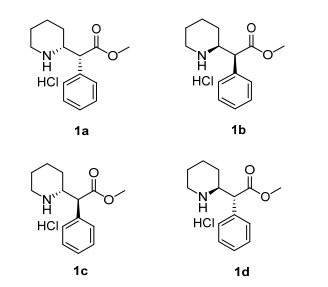
 图 1
盐酸右哌甲酯及其3个光学异构体
Figure 1.
Demethylphenidate hydrochloride and its three related optical isomers
图 1
盐酸右哌甲酯及其3个光学异构体
Figure 1.
Demethylphenidate hydrochloride and its three related optical isomers
引用本文:
张飞龙, 李伯男, 孙媛媛, 李建其. 盐酸右哌甲酯及其光学异构体的合成[J]. 有机化学,
2016, 36(9): 2162-2167.
doi:
10.6023/cjoc201603001

Citation: Zhang Feilong, Li Bonan, Sun Yuanyuana, Li Jianqi. Synthesis of Demethylphenidate Hydrochloride and Its Related Optical Isomers[J]. Chinese Journal of Organic Chemistry, 2016, 36(9): 2162-2167. doi: 10.6023/cjoc201603001
Citation: Zhang Feilong, Li Bonan, Sun Yuanyuana, Li Jianqi. Synthesis of Demethylphenidate Hydrochloride and Its Related Optical Isomers[J]. Chinese Journal of Organic Chemistry, 2016, 36(9): 2162-2167. doi: 10.6023/cjoc201603001
盐酸右哌甲酯及其光学异构体的合成
摘要:
为进行抗小儿多动症原料药盐酸右哌甲酯的质量研究,合成了盐酸右哌甲酯及3个有关光学异构体.合成工作从2-氯吡啶2和苯乙腈3出发,经两步反应生成中间体5.5经加压氢化反应生成化合物6,化合物6经叔丁醇钾构型调整得化合物7a顺式消旋体,7a经D-二苯甲酰酒石酸拆分,甲酯化成盐,得化合物1a(盐酸右哌甲酯);7a经L-二苯甲酰酒石酸拆分,甲酯化成盐,得化合物1b;化合物6经乙酸乙酯打浆重结晶得化合物7b反式消旋体,7b经D-酒石酸拆分,甲酯化成盐,得化合物1c;7b经L-酒石酸拆分,甲酯化成盐,得化合物1d.合成的4个光学异构体经MS、NMR确证结构,HPLC确证化学和光学纯度,旋光仪确定旋光,盐酸右哌甲酯1a与化合物1d经X射线单晶衍射确定绝对构型,可作为盐酸右哌甲酯质量控制过程中的光学杂质对照品.
English
Synthesis of Demethylphenidate Hydrochloride and Its Related Optical Isomers
Abstract:
For the purpose of quality control of demethylphenidate hydrochloride (1a), three related optical substances were synthesized and characterized. (S, S)-Methylphenidate hydrochloride (1b), (R, S)-methylphenidate hydrochloride (1c) and (S, R)-methylphenidate hydrochloride (1d) were synthesized via the same route with demethylphenidate hydrochloride but changing different intermediates or resolving agents. The structures of four optical isomers were tested by MS and NMR spectra, the chemical and optical purities were analyzed by HPLC spectra, the specific rotation values were also measured, the absolute configurations of compounds 1a and 1d were confirmed by X-ray study. The three optical isomers can be used as the reference substances of the related optical substances in the quality control of demethylphenidate hydrochloride.
-
Key words:
- demethylphenidate hydrochloride
- / related optical isomers
- / synthesis
-
盐酸右哌甲酯缓释胶囊用于治疗6岁及以上患者的注意缺陷多动障碍(ADHD)[1~3], 于2005年5月26日由Novartis公司[4, 5]通过美国食品药物管理局(FDA)申请上市, 商品名为Focalin XR.本品2015年美国销售额约6亿美元, 且销量呈逐年上升趋势, 市场前景非常好.2010年7月6日, Shire plc公司宣布FDA批准Daytrana (哌甲酯透皮贴剂)上市, 用于治疗13~17岁青少年的注意力缺陷/多动障碍(ADHD)[6], 这是第一个治疗ADHD的非口服药, 这种药不仅是治疗ADHD的唯一贴剂, 而且还为向那些被确诊患上ADHD的人提供了一种方便的给药选择[7, 8].到目前为止, 除了盐酸右哌甲酯本身, 其另外3个光学异构体, 尤其是一对差向异构体还没有文献就其不对称合成工作开展过完整研究, 通过相对方便的手段系统化地将其全部得到, 以对比其合成过程及化合物性质的差异.因此, 为了对上述两种药品的原料药盐酸右哌甲酯合成工艺的质量控制过程进行研究, 同时对这一类化合物的性质有一个系统的了解, 制备其相关光学异构体就产生了重要意义.
盐酸右哌甲酯作为一个临床上被证明十分有效的药用分子, 其合成制备过程一直受到广大化学及药学工作者的关注, 先后有不同的合成路线与方法被报道出来[9].其中, 诺华公司最先开发出了以不对称羟醛缩合反应为关键步骤的合成路线[10], 并在其后进行了改进[11].之后又陆续有科学家先后报道了由苯甲酰甲酸甲酯为起始原料[12, 13], 或以哌啶甲酸甲酯为起始原料[14]的合成路线.Matsumura等[15, 16]开发的不对称合成路线以Evans酰胺的偶联反应为关键步骤, 之后Fox等[17]在这条路线的基础上, 使用甲基苄胺为手性助剂, 对该路线进行了改进.为了以适用于工业化大规模生产的方法最终得到目标光学异构体, 我们最终选择一条比较简洁的合成路线[18~22], 合成了盐酸右哌甲酯及其3个光学异构体, 分别为(R, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1a), (S, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1b), (R, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1c)和(S, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1d), 其结构见图 1.
图 1
盐酸右哌甲酯及其3个光学异构体
Figure 1.
Demethylphenidate hydrochloride and its three related optical isomers
1 结果与讨论
如Scheme 1所示, 该路线从可大规模获得的商品原材料2-氯吡啶(2)和苯乙腈(3)出发, 以甲苯为溶剂经氨基钠作用生成2-吡啶-2-基苯乙腈(4), 该化合物在浓硫酸作用下氰基被水解生成2-吡啶-2-基苯乙酰胺(5).化合物5以乙酸为溶剂, 经钯碳催化剂在70 ℃条件下加压氢化, 可得2-哌啶-2-基苯乙酰胺(6).使用乙酸为溶剂有两个目的, 一是乙酸沸点相对较高, 可适应于氢化反应所需要的较高反应温度, 二是该反应无论是反应物5还是产物6由于吡啶或哌啶基团的存在都呈明显的碱性, 使用酸性溶剂有助于增强底物在溶剂中的溶解性, 以利于提高氢化反应的效率.氢化完成后的产物6是顺式和反式异构体的混合物, 多次实验发现, 化合物6中顺式和反式异构体的比例与氢化反应时采用的氢气压力有一定关系(反应条件筛选见表 1).由于吡啶环存在一定的芳香性, 该反应必须有在一定压力下, 用10%钯含量的钯碳催化剂催化才能进行, 压力小于0.3 MPa反应不能发生, 在1~5.0 MPa范围内压力越大反式产物所占比例相对越高, 顺式产物所占比例相对越低, 但总的趋势仍是反式多于顺式.
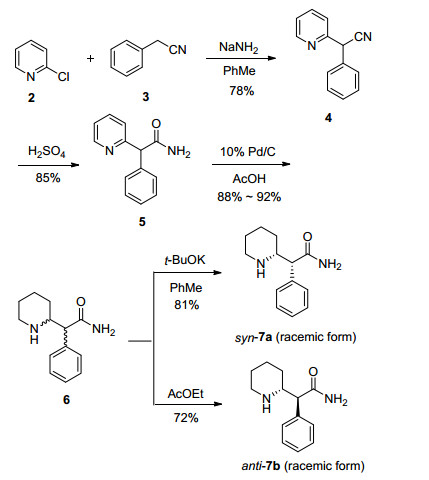 图 图式1
顺式和反式2-哌啶-2-基苯乙酰胺的合成
Figure 图式1.
Synthesis of syn-and anti-form of 2-piperidine-2-yl-phenylacetamide
图 图式1
顺式和反式2-哌啶-2-基苯乙酰胺的合成
Figure 图式1.
Synthesis of syn-and anti-form of 2-piperidine-2-yl-phenylacetamide
 表 1
化合物5氢化反应条件筛选
Table 1.
Condition screenings of hydrogenation reaction of compound 5
表 1
化合物5氢化反应条件筛选
Table 1.
Condition screenings of hydrogenation reaction of compound 5
钯碳/% t/h PHz/MPa Yielda/% 7a/7b (%)b 5 24 2.0±0.1 <5 — 10 24 <0.3 Trace — 10 15 1.0±0.1 91 25:75 10 8 2.0±0.1 90 14:86 10 4 3.0±0.3 90 9:91 10 3 5.0±0.5 88 3:97 a分离收率; bHPLC测定. 表 1 化合物5氢化反应条件筛选
Table 1. Condition screenings of hydrogenation reaction of compound 5化合物6若采用叔丁醇钾作用于酰胺α位(同时也是苯甲位)的活泼氢经进行构型调整, 由于顺式异构体相对于反式更加稳定, 经构型调整后全部得到顺式异构体7a.如将化合物6在氢化完成但尚未固化的条件下直接在乙酸乙酯内打浆处理, 则可完成类似于重结晶的纯化过程以去除相对少量的顺式异构体, 得到纯净的反式异构体7b.
盐酸右哌甲酯1a和化合物1b的合成路线如Scheme 2所示, 化合物7a经D-二苯甲酰酒石酸[19]手性拆分得到中间体8a, 8a经甲酯化和盐酸成盐得盐酸右哌甲酯1a, 两步收率23.5%;化合物7a经L-二苯甲酰酒石酸手性拆分得中间体8b, 8b经甲酯化和盐酸成盐得化合物1b, 两步收率21.6%.
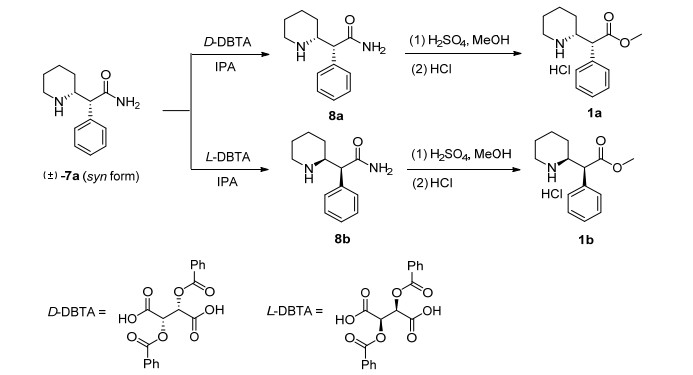 图 图式2
(R, R)-和(S, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐的合成
Figure 图式2.
Synthesis of (R, R)-and (S, S)-methyl 2-piperidine-2-yl-phenylacetate hydrochlorides
图 图式2
(R, R)-和(S, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐的合成
Figure 图式2.
Synthesis of (R, R)-and (S, S)-methyl 2-piperidine-2-yl-phenylacetate hydrochlorides
化合物1c和1d的合成路线如Scheme 3所示, 化合物7b经D-酒石酸手性拆分得中间体8c, 8c经甲酯化和盐酸成盐得化合物1c, 两步收率20.8%;化合物7b经L-酒石酸手性拆分得中间体8d, 8d经甲酯化和盐酸成盐得化合物1d, 两步收率19.1%.
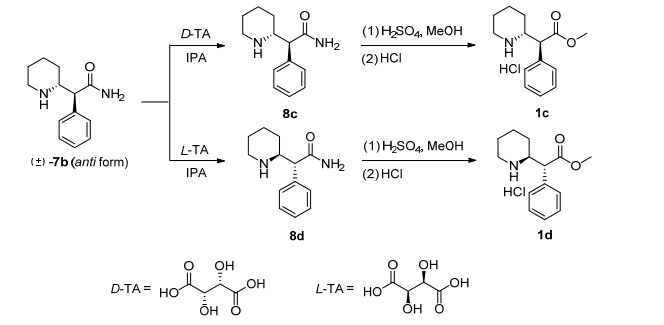 图 图式3
(R, S)-和(S, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐的合成
Figure 图式3.
Synthesis of (R, S)-and (S, R)-methyl 2-piperidine-2-yl-phenylacetate hydrochlorides
图 图式3
(R, S)-和(S, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐的合成
Figure 图式3.
Synthesis of (R, S)-and (S, R)-methyl 2-piperidine-2-yl-phenylacetate hydrochlorides
盐酸右哌甲酯1a与化合物1d经X射线单晶衍射确定绝对构型, 其晶体结构如图 2和图 3所示, 数据存于英国剑桥大学的剑桥晶体数据中心(CCDC), CCDC号分别为1453371和1453372.
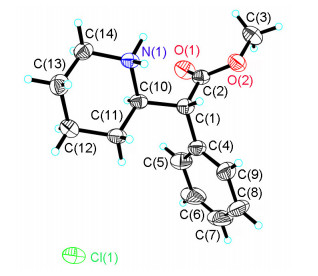 图 2
化合物1a的晶体结构
Figure 2.
Crystal structure of compound 1a
图 2
化合物1a的晶体结构
Figure 2.
Crystal structure of compound 1a
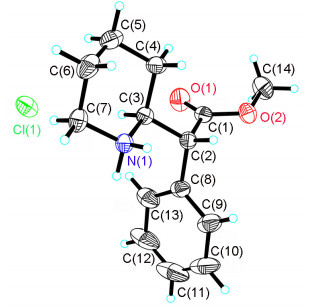 图 3
化合物1d的晶体结构
Figure 3.
Crystal structure of compound 1d
图 3
化合物1d的晶体结构
Figure 3.
Crystal structure of compound 1d
2 结论
本文按照盐酸右哌甲酯的合成工艺路线经过改变合成工艺和拆分试剂合成了盐酸右哌甲酯1a和其3个光学异构体1b, 1c和1d, 通过相对简洁的合成路线以系统化的方式得到合格的4个光学异构体样品.合成的盐酸右哌甲酯和其它三个光学异构体经MS和NMR确证结构, HPLC确证化学和光学纯度, 旋光仪确定旋光值, 盐酸右哌甲酯1a和化合物1d经X射线单晶衍射确定了绝对构型.上述光学异构体的合成方法为盐酸右哌甲酯生产过程中所需大量杂质对照品的制备奠定了基础, 亦对盐酸右哌甲酯的质量控制具有较大的意义.
3 实验部分
3.1 仪器与试剂
核磁共振谱采用Unity INOVA 400型核磁共振仪测定(DMSO-d6为溶剂, TMS为内标); 质谱采用Agilent 1946B ESI-MS型质谱仪测定; 旋转蒸发仪为Bucher型; 旋光仪为MCP 500型; 单晶X射线衍射仪为Bruker SMART APEX-Ⅱ型.实验所用试剂均为市售分析纯, 无水试剂与溶剂使用前均按要求进行预处理.
3.2 实验方法
3.2.3 (R, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐(盐酸右哌甲酯, 1a)的合成
将中间体7a (30.0 g, 137.6 mmol)和D-二苯甲酰酒石酸(51.7 g, 137.6 mmol)加入2 L四口烧瓶中, 再加入异丙醇(600 mL), 搅拌均匀后加热至55 ℃, 固体全部溶解, 保持该温度搅拌反应60 min.将溶液缓慢降温至30 ℃, 有大量固体析出并过滤.将得到的固体用异丙醇(150 mL)重结晶一遍, 然后缓慢加入6 mol/L盐酸中(300 mL), 充分搅拌反应2 h.过滤去除析出的固体, 保留滤液.将此滤液置于1 L四口烧瓶中, 缓慢滴加30%氢氧化钠水溶液至pH值到13~14为止, 有类白色固体析出, 过滤烘干得中间体8a (10.6 g).
将中间体5加入500 mL三口烧瓶中, 加甲醇(300 mL)溶解, 然后缓慢滴加浓硫酸(15 mL), 滴加完毕后加热回流反应60 h.HPLC检测反应转化完毕后, 将溶剂旋干, 加水(20 mL), 乙酸异丙酯(50 mL), 搅拌均匀后缓慢滴加30%氢氧化钠水溶液, 至水相pH值到13~14为止, 静置分液, 旋转蒸发至有机溶剂减少约90%, 缓慢滴加浓盐酸(2 mL), 瓶内析出白色固体, 过滤烘干即得到产物1a[20, 23] 8.7 g, 两步收率23.5%.HPLC纯度99.9%, ee值 > 99.9%.

3.2.4 (S, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1b)的合成
将中间体7a (30.0 g, 137.6 mmol)和L-二苯甲酰酒石酸(51.7 g, 137.6 mmol)加入2 L四口烧瓶中, 加入异丙醇(600 mL)搅拌均匀后加热至55 ℃, 固体全部溶解, 保持该温度搅拌反应60 min.将溶液缓慢降温至30 ℃, 有大量固体析出并过滤.将得到的固体用异丙醇(150 mL)重结晶一遍, 然后缓慢加入6 mol/L盐酸中(300 mL), 充分搅拌反应2 h, 过滤去除析出的固体, 保留滤液.将此滤液置于1 L四口烧瓶中, 缓慢滴加30%氢氧化钠水溶液至pH值达到14为止, 有类白色固体析出, 过滤烘干得中间体8b (10.1g).
将中间体8b加入500 mL三口烧瓶中, 加甲醇(300 mL)溶解, 然后缓慢滴加浓硫酸(15 mL), 滴加完毕后加热回流反应60 h.HPLC检测反应转化完毕后, 将溶剂旋干, 加水(20 mL)、乙酸异丙酯(50 mL), 搅拌均匀后缓慢滴加30%氢氧化钠水溶液至水相pH到13~14为止, 静置分液, 旋转蒸发至有机溶剂减少约90%, 缓慢滴加浓盐酸(2 mL), 瓶内析出白色固体, 过滤烘干即得到产物1b 8.0 g, 两步收率21.6%.HPLC纯度97.7%, ee值 > 99.9%.
3.2.1 顺式-2-哌啶-2-基苯乙酰胺(7a)的合成
在容积为2 L高压氢化反应釜内加入化合物5 (100.0 g, 471.7 mmol), 10%钯碳(10 g)、乙酸(1 L), 用氢气钢瓶加压至3.0 MPa, 加热至70 ℃开始反应.4 h后冷却降温释放压力, 将钯碳催化剂过滤, 溶剂乙酸旋干, 加入水(1 L), 将溶液降温至10 ℃, 然后在快速搅拌下缓慢滴加30%氢氧化钠水溶液, 滴加至有大量固体析出, 溶液pH值为13~14为止.过滤收集固体, 并将固体烘干, 此时得到中间体6 (89.3 g).
在5 L四口烧瓶中加入中间体6和甲苯(2.7 L), 搅拌均匀后加入叔丁醇钾(89 g, 794.6 mmol), 加热至70 ℃反应.16 h后将反应降至室温, 缓慢滴加水(500 mL)以淬灭剩余的叔丁醇钾, 然后缓慢滴加6 mol•L-1盐酸(500 mL), 滴毕后快速搅拌, 使有机相与水相充分接触, 静置分层, 分液取水相.在分离得到的水相中缓慢滴加30%氢氧化钠水溶液, 直至溶液的pH值到13~14为止, 此时有大量固体析出, 过滤收集固体并烘干, 得浅黄色固体70.1 g, 两步收率68.2%.
3.2.1 2-吡啶-2-基苯乙酰胺(5)的合成
在5 L的四口瓶中加入甲苯1.5 L, 室温搅拌下通入氮气吹洗0.5 h, 然后加入氨基钠(293.0 g, 7.51 mol).冰浴使体系温度降为10 ℃, 搅拌下缓慢滴加苯乙腈(570.0 g, 4.87 mol), 随后滴加2-氯吡啶(500.0 g, 4.42 mol), 保持体系温度低于30 ℃.滴毕撤离冰浴, 常温搅拌反应4 h.反应结束后缓慢加水(200 mL)淬灭过量的氨基钠, 并用水(200 mL)洗涤有机相3次.将有机相中的甲苯蒸出约80%, 然后加入石油醚(200 mL), 搅拌均匀后用冰浴冷却(1 h), 产物从母液中析出, 过滤并烘干得化合物4, 为浅黄色固体668.1 g, 收率78%.
在3 L的四口瓶中加入浓硫酸(400 mL), 冰浴冷至10 ℃, 搅拌下分批加入上一步产物(300 g, 1.55 mol), 保持反应温度低于20 ℃, 加毕后升温至室温反应, 16 h后冰浴冷至10 ℃滴加水(1.5 L), 再使用30% NaOH水溶液调pH至13~14, 抽滤并干燥, 得淡黄色固体278.5 g, 收率85%.
3.2.6 (S, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1d)的合成
将中间体7b (30.0 g, 137.6 mmol)和L-酒石酸(20.6 g, 137.6 mmol)加入2 L四口烧瓶中, 加入异丙醇(600 mL)搅拌均匀后加热至55 ℃, 固体全部溶解, 保持该温度搅拌反应60 min, 将溶液缓慢降温至30 ℃, 有大量固体析出并过滤.将此固体溶解于水中(500 mL), 缓慢滴加50%氢氧化钠水溶液至pH值到13~14为止, 有类白色固体析出, 过滤烘干得中间体8d (9.0 g).
将中间体8d加入500 mL三口烧瓶中, 加甲醇(300 mL)溶解, 然后缓慢滴加浓硫酸(15 mL), 滴加完毕后加热回流反应60 h.HPLC检测反应转化完毕后, 将溶剂旋干, 加水(20 mL)、乙酸异丙酯(50 mL), 搅拌均匀后缓慢滴加30%氢氧化钠水溶液至水相pH值到13~14为止.静置分液, 旋转蒸发至有机溶剂减少约90%, 缓慢滴加浓盐酸(2 mL), 瓶内析出白色固体, 过滤烘干即得到产物1d 7.1 g, 两步收率19.1%.HPLC纯度96.6%, ee值 > 99.2%.
辅助材料(Supporting Information) 中间体及最终产物的HPLC谱图及化合物1a和1d的晶体表征数据.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
3.2.5 (R, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐(1c)的合成
将中间体7b (30.0 g, 137.6 mmol)和D-酒石酸(20.6 g, 137.6 mmol)加入2 L四口烧瓶中, 加入异丙醇(600 mL)搅拌均匀后加热至55 ℃, 固体全部溶解, 保持该温度搅拌反应60 min, 将溶液缓慢降温至30 ℃, 有大量固体析出并过滤.将此固体溶解于水中(500 mL), 缓慢滴加50%氢氧化钠水溶液至pH值到13~14为止, 有类白色固体析出, 过滤烘干得中间体8c (9.9 g).
将中间体8c加入500 mL三口烧瓶中, 加甲醇(300 mL)溶解, 然后缓慢滴加浓硫酸(15 mL), 滴加完毕后加热回流反应60 h.HPLC检测反应转化完毕后, 将溶剂旋干, 加水(20 mL)、乙酸异丙酯(50 mL), 搅拌均匀后缓慢滴加30%氢氧化钠水溶液至水相pH值到13~14为止.静置分液, 旋转蒸发至有机溶剂减少约90%, 缓慢滴加浓盐酸(2 mL), 瓶内析出白色固体, 过滤烘干即得到产物1c 7.7 g, 收率20.8%.HPLC纯度98.1%, ee值 > 99.9%.
3.2.2 反式-2-哌啶-2-基苯乙酰胺(7b)的合成
在容积为2 L高压氢化反应釜内加入化合物5 (100.0 g, 471.7 mmol)、10%钯碳(10 g)、乙酸(1 L), 用氢气钢瓶加压至3.0 MPa, 加热至70 ℃开始反应.4 h后冷却降温释放压力, 将钯碳过滤, 溶剂乙酸旋干, 此时得到的是浅黄色粘油.在此粘油中加入乙酸乙酯(1 L), 搅拌下逐渐有固体析出, 将固体收集过滤, 然后溶解于水中(800 mL), 缓慢滴加30%氢氧化钠水溶液, 直至溶液pH至13~14为止, 有大量固体析出, 过滤并烘干, 得到浅黄色固体62.3 g, 收率60.6%.
-
-
[1]
Volkmar, F. R. Am. J. Psychiatry 2003, 160, 1025. doi: 10.1176/appi.ajp.160.6.1025
-
[2]
静进, 中国实用儿科临床杂志, 2012, 27, 965. http://d.wanfangdata.com.cn/Periodical/syeklczz201212026Jing, J. Chin. J. App. Clin. Pediatrics 2012, 27, 965(in Chinese). http://d.wanfangdata.com.cn/Periodical/syeklczz201212026
-
[3]
刘津, 王玉凤, 中华神经科杂志, 2001, 34, 247. http://d.wanfangdata.com.cn/Periodical_zhjsk200104024.aspxLiu, J.; Wang, Y. F. Chin. J. Neurology 2001, 34, 247(in Chi-nese). http://d.wanfangdata.com.cn/Periodical_zhjsk200104024.aspx
-
[4]
邹丽萍, 世界临床药物, 2005, 26, 357. http://doi.med.wanfangdata.com.cn/qk/gwyy-hcy200506009Zou, L. P. World Clin. Drugs 2005, 26, 357(in Chinese). http://doi.med.wanfangdata.com.cn/qk/gwyy-hcy200506009
-
[5]
Har, D.; Prashad, M. US 6100401, 2000[Chem. Abstr. 2000, 133, 150475].
-
[6]
李瑛, 吴佳, 中国药师, 2007, 6, 601. http://www.cqvip.com/Main/Detail.aspx?id=24576016Li, Y.; Wu, J. China Pharmacist 2007, 6, 601(in Chinese). http://www.cqvip.com/Main/Detail.aspx?id=24576016
-
[7]
Biederman, J.; Wilens, T.; Mick, E.; Spencer, T.; Faraone, S. V.; Pediatrics 1999, 104, 20. doi: 10.1542/peds.104.2.e20
-
[8]
Srinivas, N.; Nuggehally, R. Clin. Pharm. Therap. 1992, 52, 561. doi: 10.1038/clpt.1992.185
-
[9]
Prashad, M. Adv. Synth. Catal. 2001, 343, 379 doi: 10.1002/(ISSN)1615-4169
-
[10]
Prashad, M.; Kim, H. Y.; Lu, Y.; Liu, Y.; Har, D.; Repic, O.; Blacklock, T. J.; Giannousis, P. J. Org. Chem. 1999, 64, 1750. doi: 10.1021/jo9821473
-
[11]
Prashad, M.; Liu, Y.; Kim, H. Y.; Repic, O.; Blacklock, T. J. Tetrahedron Asymmetry 1999, 10, 3479. doi: 10.1016/S0957-4166(99)00384-5
-
[12]
Gutman, A.; Zaltsman, I.; Shalimov, A.; Sotrihin, M.; Nisnevich, G.; Yudovich, L.; Fedotev, I. US 20040180928, 2004[Chem. Abstr. 2004, 141, 277499].
-
[13]
吴增, 余永强, 朱雍, 陆涛, 海峡药学, 2010, 22, 211.Wu, Z.; Yu, Y. Q.; Zhu, Y.; Lu, T. Strait Pharm. J. 2010, 22, 211(in Chinese).
-
[14]
Davies, H. M. L.; Hansen, T.; Hopper, D. W.; Panaro, S. A. J. Am. Chem. Soc. 1999, 121, 6509. doi: 10.1021/ja9910715
-
[15]
Matsumura, Y.; Kanda, Y.; Shirai, K.; Onomura, O.; Maki, T. Org. Lett. 1999, 1, 175. doi: 10.1021/ol9905046
-
[16]
Matsumura, Y.; Kanda, Y.; Shirai, K.; Onomura, O.; Maki, T. Tetrahedron 2000, 56, 7411. doi: 10.1016/S0040-4020(00)00653-0
-
[17]
Fox, M. E.; Paul, J. M. WO 9735836, 1997[Chem. Abstr. 1997, 127, 247922].
-
[18]
Axten J. M.; Krim, L.; Kung, H. F.; Winkler, J. D. J. Org. Chem. 1998, 63, 9628. doi: 10.1021/jo982214t
-
[19]
Chan, A. B.; Gundecha S. S.; Kadam, P. N.; Maikap, G. C.; Gurjar, M. K. Org. Process Res. Dev. 2010, 14, 1473. doi: 10.1021/op100197g
-
[20]
Prashad, M.; Hu, B.; Repic, O.; Blacklock, T. J.; Giannousis P. Org. Process Res. Dev. 2000, 4, 55. doi: 10.1021/op990195z
-
[21]
Kumar, A.; Singh, D.; Patil, S. H.; Mahale, G. D.; Sawant, U. A. US 20050277667, 2005[Chem. Abstr. 2005, 144, 51454].
-
[22]
Hu, B.; Prashad, M. US 6162919, 2000[Chem. Abstr. 2000, 134, 42069].
-
[1]
-

图 1 盐酸右哌甲酯及其3个光学异构体
Figure 1 Demethylphenidate hydrochloride and its three related optical isomers

图式1 顺式和反式2-哌啶-2-基苯乙酰胺的合成
Scheme 1 Synthesis of syn-and anti-form of 2-piperidine-2-yl-phenylacetamide

图式2 (R, R)-和(S, S)-2-哌啶-2-基苯乙酸甲酯盐酸盐的合成
Scheme 2 Synthesis of (R, R)-and (S, S)-methyl 2-piperidine-2-yl-phenylacetate hydrochlorides

图式3 (R, S)-和(S, R)-2-哌啶-2-基苯乙酸甲酯盐酸盐的合成
Scheme 3 Synthesis of (R, S)-and (S, R)-methyl 2-piperidine-2-yl-phenylacetate hydrochlorides
表 1 化合物5氢化反应条件筛选
Table 1. Condition screenings of hydrogenation reaction of compound 5
钯碳/% t/h PHz/MPa Yielda/% 7a/7b (%)b 5 24 2.0±0.1 <5 — 10 24 <0.3 Trace — 10 15 1.0±0.1 91 25:75 10 8 2.0±0.1 90 14:86 10 4 3.0±0.3 90 9:91 10 3 5.0±0.5 88 3:97 a分离收率; bHPLC测定.  下载: 导出CSV
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 6233
- HTML全文浏览量: 2720
 下载:
下载:
