
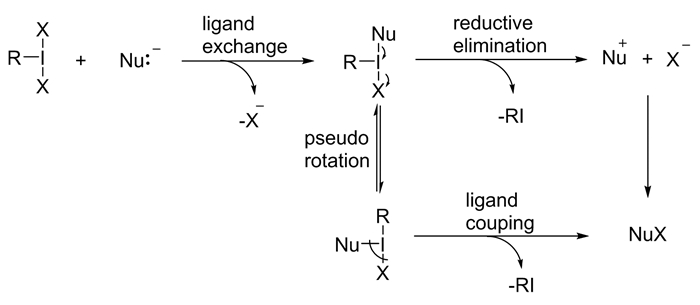
图式 1.
λ3-碘烷的典型结构
Scheme 1.
Typical structures of λ3-iodanes

三价碘试剂,被国际纯粹与应用化学联合会(IUPAC)命名为“λ3-碘烷”(图式 1)。三价碘试剂RIX2,其碘原子有10个电子,从三维空间来看是一个三角双锥型分子,碘原子处在中心,取代基R和两对孤对电子位于平面位置,两个杂原子的配体X位于顶点,分子中的I-X键长比普通的I-X键长长。碘盐的结构与RIX2类似,其R-I-R键角接近90°。同时,“λ3-碘烷”RIX2中的成键轨道被碘原子非杂化的5p轨道的2个电子和两个配体X的2个电子所占据,这种结构使得三价碘的化学键比普通的共价键更长、更弱和更容易极化,致使三价碘化合物具有较高的亲电活性。
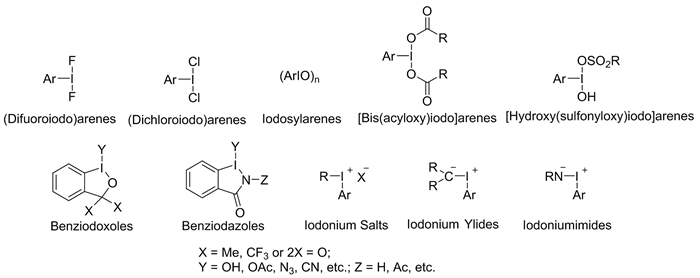
三价碘试剂通常按照与碘原子配位的配体类型进行分类,图式 2列出了在有机合成中应用极为广泛的典型三价碘试剂。其中,醋酸碘苯PhI(OAc)2是二羧酸芳基碘类试剂中最重要的并且已商业化生产的代表性试剂;羟基甲苯磺酰碘苯PhI(OH)OTs是最具代表性的有机磺酸基芳基碘类试剂,目前也已商业化生产,其缩写为HTIB,通常称为Koser试剂。
三价碘试剂常被视为氧化剂和亲电试剂而广泛应用于有机合成当中,其三个典型的反应过程为[1]:(1)配体交换;(2)还原消除;(3)配体偶联(图式 3)。除此之外,在一定的条件下,三价碘试剂可以发生诸如单电子转移的自由基类型的反应。本文就近年来三价碘试剂促进的氧化反应进行综述。
醋酸碘苯等二羧酸芳基碘类化合物可以作为醇的氧化剂[2]。例如,醋酸碘苯和碘的体系可以有效地将仲醇氧化为酮,将伯醇氧化为羧酸。另外,伯醇1可以在甲醇作溶剂的醋酸碘苯和碘的体系中反应,以很高的产率得到羧酸甲酯类化合物2(式(1))[3]。3-硝基醋酸碘苯(4)和三氟化硼乙醚体系促进的醇氧化也有报道,反应的机理可能是首先3-硝基醋酸碘苯和醇(3)之间进行了快速配体交换生成烷氧基碘的中间体5,后者经过还原消除3-硝基碘苯生成酮类化合物6(式(2))[4]。
|
|
(1) |
|
|
(2) |
醋酸碘苯和四甲基哌啶氮氧化物(TEMPO)体系也是醇类化合物非常高效的氧化体系,这种体系可以将各种伯醇和仲醇氧化为相应的羰基化合物[5]。值得一提的是,在醋酸碘苯-TEMPO体系中,伯醇可以选择性地氧化为醛,而不会进一步氧化生成羧酸,即使醇分子中有易被氧化的双键存在时,双键也不会受到影响。例如,橙花醇7在催化量TEMPO存在下,被醋酸碘苯在水和乙腈混合溶剂中以高产率氧化为醛类化合物8(式(3))[6]。
|
|
(3) |
二氯芳基碘除了作为氯化试剂之外,也可以用于醇的氧化。例如,仲醇类化合物在二氯芳基碘、叠氮化钠的作用下能够以很高的产率合成相应的酮类化合物[7]。然而,伯醇9在二氯碘苯和叠氮化钠的体系中,经过Curtius重排主要生成酰基叠氮化合物10,这样就可以利用伯醇和胺在PhICl2-NaN3体系中反应一锅法来合成各种脲的衍生物12,这为脲衍生物的合成提供了一种有效的方法[8]。(式(4))。
|
|
(4) |
亚碘酰苯也可以作为醇的氧化剂,特别是在溴化钾催化下,亚碘酰苯可以将各种伯醇和仲醇氧化,即使醇分子中存在敏感的官能团如醚键、酯基、磺酰胺基以及叠氮基,都不受影响[9]。通常,伯醇在亚碘酰苯和溴化钾体系中氧化生成羧酸(式(5)),各种仲醇在该体系中氧化生成相应的酮类化合物。
|
|
(5) |
三价碘试剂是羰基化合物氧化官能团化的常用试剂,特别是生成的具有α位官能团化的物质普遍存在于天然产物如聚酮化合物、萜类化合物以及生物碱中,所以受到有机化学家的强烈关注。在20世纪80年代,有机化学家就利用亚碘酰苯、醋酸碘苯以及其他三价碘试剂进行了羰基化合物的氧化官能团化。其中,醋酸碘苯、氢氧化钾和甲醇是酮类化合物氧化官能团化特别有效的体系,这种体系已经广泛用于酮、酯等其他羰基化合物的氧化官能团化(式(6))[10]。
二氟芳基碘是非常有效的选择性氟化试剂,也常用于羰基化合物如β-酮酯和β-二羰基衍生物α位的选择性氟化[11]。特别是,诸如β-酮酯、β-酮酰胺、β-二酮类化合物,在温和条件下与二氟芳基碘反应均能生成羰基α位单氟代的产物(式(7))[12]。
|
|
(6) |
|
|
(7) |
α-苯基硫乙酰胺20也能与二氟芳基碘化合物在温和条件下反应,生成羰基α位氟化的产物21,同样,α-苯基硫乙酰酯22在类似条件下也能够生成相应的氟化产物23(式(8))[13]。
|
|
(8) |
二氟芳基碘促进的羰基α位二氟化反应也有报道。例如,Murphy等采用底物24与4-二氟化碘甲苯反应,生成相应的二氟代产物25,这是一种无金属催化的用来高效地实现羰基α位二氟化的重要方法(式(9))[14]。
|
|
(9) |
二氯芳基碘是一种高效的氯化试剂,其反应模式有两种,一是在光化学或自由基引发剂存在下所发生的自由基过程;二是亲电加成过程。二氯芳基碘经常用于酮α位sp3-碳的氯化[15]。无论是脂肪族酮还是芳香族酮,在二氯芳基碘的作用下,均能直接转变成相应的α位氯代产物,其机理可以是自由基过程也可以是离子型过程,主要取决于反应的条件。二氯芳基碘促进的β-酮酯α位对映选择性氯化也有报道。例如,β-酮酯26在手性金属催化剂钛配合物(27)的催化下,在甲苯溶剂中与二氯碘甲苯反应,能够生成相应的α位氯代产物28,唯一不足的是,反应的产率适中,对映选择性相对较低(式(10))[16]。
|
|
(10) |
另外,羟基磺酰氧基芳基碘也可以促进羰基化合物α位的官能团化。例如,Koser试剂和酮反应生成α位被对甲苯磺酰基取代的酮,这是一类化学选择性非常高的反应,不同的官能团如芳环和双键在该反应条件下耐受性很强,式(11)就是利用Koser试剂实现了去毒毒素生物碱羰基α位的氧化官能团化[17]。
|
|
(11) |
三价碘试剂作为高效的氧化剂,广泛用于烯烃的氧化[18]。例如,烯烃和双三氟乙酸碘苯反应,生成邻位双三氟乙酰氧基的衍生物,经水解作用生成乙二醇或羰基化合物[19]。一个典型的例子是,环己烯31和三氟乙酸碘苯在二氯甲烷中回流,生成顺式的1, 2-双三氟乙酰氧基衍生物32;双环烯烃1, 4-二氢-1, 4-甲桥萘33和三氟乙酸碘苯在类似条件下以95%的产率得到氧化产物34。类似的反应也能在强酸的条件下发生(式(12))[20]。
|
|
(12) |
烯烃的对应选择性双乙酰氧基化可以在醋酸碘苯和三氟化硼乙醚体系中进行,在这种体系中,烯烃和醋酸碘苯反应可以选择性地生成syn或anti型双乙酰氧基化产物,产物的选择性主要取决于体系中是否有水存在,当体系中含有水时,主要生成syn产物,反之生成anti产物[21]。各种烯烃如芳香族烯烃、脂肪族烯烃、环状烯烃、α, β不饱和酯在这一体系中均能生成相应的邻双乙酰氧基化产物,反应的机理可能是生成重要的碳正离子中间体38(式(13))[22]。
|
|
(13) |
另外,缺电子烯烃在三价碘试剂的促进下能够发生氧化溴氨基化反应。例如,Wang等[23]报道了醋酸碘苯促进的α, β不饱和类化合物40的区域、立体选择性溴氨基化反应(式(14))。
|
|
(14) |
Kirschning等[24]报道了醋酸碘苯促进的含溴或碘阴离子的物质与烯烃的立体选择性溴乙酰化或碘乙酰化反应,反应的亲电中间体二乙酰卤负离子(AcO)2X-(X=Br or I)是由相应的卤阴离子和高分子固载的高碘试剂反应得到。此外,醋酸碘苯和碘促进的亲核试剂与烯烃的反应常用来合成β-官能团化碘代烷烃。例如,在二氯甲烷中,烯烃43和氨基酸衍生物的三价高碘试剂42在四苯基碘化膦的作用下生成碘代烃44(式(15))[25]。
|
|
(15) |
Nicolaou等[26]报道了二聚金鸡纳生物碱作为手性催化剂的烯丙基醇对映选择性1, 2-二氯化反应。用反式肉桂醇45与二氯芳基碘化合物4-Ph(C6H4)ICl2,在催化量手性催化剂金鸡纳生物碱(DHQ)2PHAL 46的作用下反应,能够以很高的产率和对映选择性生成相应的二氯代产物47(式(16))。
|
|
(16) |
烯烃α位氯化反应也有报道。例如,采用取代的烯烃底物48和50,分别与二氯碘苯在N, N二甲基甲酰胺中室温反应,生成相应的氯代产物49和51,这为α位氯代烯烃类化合物的合成提供了一种简便的方法(式(17))[27]。
三价碘试剂促进的炔烃氧化反应也有报道。
|
|
(17) |
例如,Koser试剂与炔烃52反应生成炔基碘盐53和烯基碘盐54两个产物,而且,当末端炔烃取代基空间位阻较大时,如叔丁基、仲丁基、环己基和芳基等,炔基碘盐53便成为主要产物;当末端炔烃取代基没有立体障碍时,主要生成烯基碘盐54(式(18))[28]。
|
|
(18) |
三价碘试剂可以用于苯酚、萘酚等酚类化合物的氧化硫氰化反应。例如,苯酚衍生物55在PhICl2-Pb(SCN)2体系中生成对位硫氰化产物56(式(19))[29]。
|
|
(19) |
Ciufolini等[30]报道了将酚类物质57氧化为螺环内酰胺物质的方法,他们用醋酸碘苯作为氧化剂,三氟乙醇为溶剂,30min内高产率地得到了螺环内酰胺物质58(式(20))。
|
|
(20) |
Sabot等[31]也报道了一种将酚羟基氧化为酮的方法,反应利用醋酸碘苯作为氧化剂使酚类衍生物59与烯丙基三甲基硅烷发生分子间氧化烯丙基化反应,生成目标产物60(式(21))。
|
|
(21) |
苄位或烯丙位C-H键的氧化可以通过醋酸碘苯和碘的体系或者是醋酸碘苯和过氧化物的体系来实现。例如,化合物61可以通过醋酸碘苯和碘的体系在对甲苯磺酰胺或4-硝基对甲苯磺酰的催化下,以1, 2-二氯乙烷为溶剂、60℃下氧化为乙酰氧基化的产物62;取代的环己烯衍生物63在醋酸碘苯和过氧化叔丁醇的体系中反应,得到烯丙位氧化的产物64。这类反应都是自由基反应(式(22))[32]。
|
|
(22) |
三氟乙酸碘苯可以促进环丙烷发生氧化裂解反应。例如,环丙烷衍生物65在三氟乙酸碘苯的氧化下反应,生成氧化裂解产物66,后者可进一步转化为天然生物碱松里汀(式(23))[33]。
|
|
(23) |
芳环的氧化可以通过三氟乙酸碘苯和Lewis酸体系来实现。例如,各种取代苯胺67在三氟乙酸碘苯和三氟化硼乙醚体系中,以乙酸或甲醇为溶剂,室温反应就能得到相应对位取代的氧化产物68和69;同样,在对甲苯磺酸作用下,取代苯胺67在该体系中室温反应就能得到对位取代的对甲苯磺酰化产物70;此外,取代苯胺67在三氟乙酸碘苯作为氧化剂、三氟甲磺酸银催化下反应生成对位取代的三氟甲磺酸化产物71(式(24))[34]。
|
|
(24) |
醋酸碘苯和三乙酸碘苯是Hofmann重排反应非常有效的试剂,无论是脂肪族甲酰胺,还是芳香族甲酰胺都能在它们的促进下发生重排,生成氨基甲酸酯类化合物74或者胺类化合物75,反应都是通过异氰酸酯中间体73来进行的(式(25))[35]。
|
|
(25) |
Kalesse等[36]报道了对称的脲衍生物的合成。反应首先是底物甲酰胺76在醋酸碘苯的促进下发生Hofmann重排,然后对重排产物进行了亲核加成得到目标产物77(式(26))。
|
|
(26) |
羟基磺酰氧基芳基碘也是氧化重排反应的有效试剂。例如,Koser试剂可以作为酰胺或胺类化合物Hofmann重排的有效氧化剂。Togo等[37]报道了利用原位生成的Koser试剂氧化脂肪族或芳香族酰亚胺发生类似Hofmann重排反应,生成相应的氨基酸产物;Koser试剂也可以促进烯烃发生氧化重排,例如,Justik等报道了Koser试剂促进的芳基烯烃78氧化重排,生成α-芳基酮79(式(27))。反应具有很好的区域选择性,这一方法也适用于环状或链状的烯烃底物,通常用于制备环状物质。
|
|
(27) |
三价碘试剂促进的Claisen重排反应是合成邻位碘代烃类化合物的重要方法。例如,Khatri等[38]报道了邻位烯丙基碘代芳烃81和82的合成,反应是通过3-甲氧基醋酸碘苯和烯丙基三甲基硅烷80发生碘的Claisen重排反应得到,该方法也成功地用于抗真菌天然产物broussin的全合成中(式(28))。
|
|
(28) |
三价碘试剂促进的缩环、扩环重排反应也有大量报道。例如,手性环醚84的合成是通过醋酸碘甲苯促进的扩环反应获得(式(29))[39]。
|
|
(29) |
醋酸碘苯可促进环丁醇衍生物85在三氟甲磺酸三甲基硅酯的作用下发生氧化缩环,生成苯基环丙基甲酮86(式(30))[40]。
|
|
(30) |
Koser试剂也可以用以促进扩环反应。例如,不饱和的三甲基硅醚87在过量Koser试剂作用下生成扩环产物苯并环庚酮衍生物88(式(31))[41]。
|
|
(31) |
三价碘试剂可以促进对位或邻位取代的酚类化合物发生去芳构化反应,产物随之在亲核试剂诸如水、醇、氟阴离子、羧酸、酰胺、肟等进攻下生成相应的环己二烯酮91和93。机理首先是底物89和三价碘试剂PhIX2进行配体交换得到中间体90,随后进行还原消除和亲核加成得到最终产物(式(32))[42]。
|
|
(32) |
Guerard等[43]报道了醋酸碘苯促进的串联重排反应,这为酚类化合物的氧化去芳构化提供了新的途径。例如,醋酸碘苯促进的Wagner-Meerwein重排,通过酚类化合物中官能团如烯丙基、芳基和烷基的迁移,最终生成氧化去芳构化的产物二酮类化合物96(式(33)),其中烯丙基迁移能够得到构型保持的立体专一性产物;此外,醋酸碘苯促进的氧化重排也能够用来快速合成含有季碳中心的官能团化的二烯酮类化合物98,这一方法在天然生物碱sceletenone的全合成中得到了应用(式(34))[44]。
|
|
(33) |
|
|
(34) |
Kita等[45]报道了酚类化合物99和醋酸碘苯在HFIP和水的体系中进行Domino反应生成螺环己二烯酮内脂100。反应的机理包括4步,首先底物99与醋酸碘苯发生配体交换生成中间体101;接着中间体101发生重排生成环己烯二酮的螺环中间体102;然后,体系中的水对中间体102的酮羰基进行亲核加成,随后在醋酸碘苯作用下,生成中间体103;最后,中间体103进行分子内环化得到目标产物螺环己二烯酮的内脂100(式(35))。
|
|
(35) |
三价碘试剂可以促进富电子芳烃在极性非亲核溶剂中发生氧化偶联反应,反应是通过单电子转移机理进行的。例如,Kita等[46]报道了芳基醚衍生物104在三氟乙酸碘苯促进下与亲核试剂的氧化偶联反应,反应首先生成碳正离子自由基中间体(105),并通过ESR进行了检测,随后在亲核试剂的进攻下生成去芳构化产物106或氧化偶联产物107(式(36))。
|
|
(36) |
另外一个例子是酚醚类化合物108和109发生的氧化交叉偶联反应,生成双芳基化合物110,此化合物可以用来构建苯并菲叮的骨架(式(37))[47]。
|
|
(37) |
后来,Kita等[48]又报道了三氟乙酸碘苯促进的芳烃111自身偶联,生成双芳基类化合物112;在相同条件下,分子间氧化交叉偶联也能发生,例如,萘113和五甲基取代苯114在三氟乙酸碘苯的促进下反应生成不对称双芳基类化合物115,而芳烃自身氧化偶联产物没有生成,这一方法可以用来制备各种二芳基化合物(式(38))。
|
|
(38) |
在后续的研究当中,Kita等又报道了三价碘试剂促进的各种分子内或分子间氧化偶联反应。其中,分子间氧化偶联反应,可以从含有N3-、AcO-、ArS-、SCN-的亲核试剂及β-二羰基化合物来制备各种取代环己二烯酮衍生物;通过分子内氧化偶联反应可以高效地合成碳环和杂环类化合物,例如,最近报道的四甲氧基取代的苯乙酸苯酚酯116的氧化偶联[49]、N-保护的吲哚类衍生物118的氧化偶联[50]以及苯乙基苄基硫醚120的氧化偶联[51]。其中氧化偶联产物119和121已经用于具有细胞毒性的Makaluvamine F的全合成中(式(39))。
|
|
(39) |
同样,噻吩类化合物也能在三价碘试剂的促进下与亲核试剂发生氧化交叉和自身偶联反应。例如,3-己基噻吩122在三氟乙酸碘苯的促进下,与TMSX(Cl、Br、SCN、CN)在二氯甲烷溶剂中室温反应,就能得到相应的交叉偶联产物123~125(式(40))[52]。
|
|
(40) |
然而,在三氟乙酸碘2-甲苯和TMSOTf的作用下,3-己基噻吩122在-78℃下于二氯甲烷中反应,便生成自身氧化偶联产物126和127,其比例为81:19(式(41))[53],除此之外,吡咯和萘酚类化合物在三价高碘试剂的促进下也能发生自身和交叉氧化偶联反应[54]。
|
|
(41) |
Koser试剂也可以用以分子内C-C键的氧化交叉偶联发生。例如,Kita等[55]报道了Koser试剂促进的杂环化合物氧化交叉偶联,为各种双芳基类化合物的合成提供了新途径。反应是以取代噻吩128和甲氧基取代的萘129在Koser试剂作为氧化剂、三甲基溴硅烷为催化剂在六氟异丙醇中进行的,室温反应3h便生成氧化交叉偶联产物130(式(42))。
|
|
(42) |
氟硼荧131也能在三氟乙酸碘苯和三氟化硼乙醚的促进下发生自身氧化偶联,生成氟硼荧二聚体132(式(43))。由于氟硼荧具有非常优越的光学性能,近年来,在染料工业中应用越来越广泛[56]。
|
|
(43) |
三价碘试剂可以促进烃类化合物和芳香醛发生氧化氨基化[57],特别是三价碘的酰胺和亚胺试剂可以有效地向分子中引入氨基官能团。ArI(OAc)NTs2和ArI(NTs2)2很容易通过双羧酸芳基碘和HNTs2原位生成,不经过分离,直接用于烯烃的氧化氨基化反应。例如,邻二胺135可以通过醋酸碘苯和双磺酰亚胺体系促进的烯烃133直接双氨基化反应来制备(式(44))[58]。
|
|
(44) |
三价碘试剂可以促进醛的氧化叠氮化反应。例如,各种芳香醛136在醋酸碘苯的促进下,与叠氮化钠在二氯甲烷中室温反应1~2 h,便得到氧化叠氮化的产物137(式(45))[59]。
|
|
(45) |
三价碘试剂可以促进烯烃进行氧化硫氰化反应。例如,取代烯烃类化合物138和硫氰化钾在醋酸碘苯的促进下反应,生成双官能团化(乙酰氧基化和硫氰化)产物139(式(46)),产物具有很好的立体选择性和区域选择性[60]。
|
|
(46) |
三价碘试剂也可以促进烯烃的氧化双硫氰化反应。例如,取代烯烃类化合物140和异硫氢酸三甲基硅酯在醋酸碘苯的促进下反应,生成双硫氰化产物141(式(47));在相同条件下,环己烯和1-甲基环己烯也可发生双硫氰化反应,并且得到反式加成产物[61]。
|
|
(47) |
三价碘试剂促进的烯烃与二芳基硒的硒芳基化反应也有报道。例如,环己烯142与二苯基硒在醋酸碘苯促进下,乙腈溶剂中40℃反应,以62%的产率得到立体选择性反式加成产物143(式(48))[62]。
|
|
(48) |
三价碘试剂也可促进醛α-位硒芳基化反应。例如,醛类化合物144与二苯基硒在醋酸碘苯的促进下,在手性催化剂145催化下,以50%~70%的产率和98%~99% ee值对映选择性地生成硒芳基醇146(式(49))[63]。通过这种方法可以制备具有生物活性的分子骨架,如β-羟基醇、α-氨基酸以及α-羟基酯类化合物。
|
|
(49) |
含有氧族(氧、硫、硒、碲)元素和氮族(氮、磷、砷、锑、铋)元素的化合物很容易被二羧酸芳基碘氧化生成相应的氧化产物。例如,二碲化合物147可以被三氟乙酸碘苯室温氧化[64];三芳基的铋类化合物149同样可以被醋酸碘苯室温氧化,生成高价态的铋化合物(式(50))[65]。
|
|
(50) |
三价碘试剂是高价碘试剂中重要的一类,被广泛地应用到现代有机合成当中,其最主要应用是作为一种高效的氧化剂,用于各种诸如C-C、C-N、C-O及杂原子间化学键的形成。三价碘试剂的特性类似于过渡金属,然而又具有过渡金属无法比拟的优点,例如,三价碘试剂容易制备、环境友好、价格低廉,因此,无论在学术研究,还是在工业领域都具有广泛的用途。
T Wirth. Hypervalent Iodine Chemistry:Modern Developments in Organic Synthesis, Topics in Current Chemistry, 2003, vol. 224, Springer, Berlin.
J Eljo, M S Carle, G K Murphy. Synlett, 2017, 28(20):2871~2875. doi: 10.1055/s-0036-1589069
N N Karade, V H Budhewar, A N Katkar et al. Arkivoc, 2006, (11):162~167.
M Kida, T Sueda, S Goto et al. Chem. Commun., 1996, 16:1933~1934.
A De Mico, R Margarita, L Parlanti et al. J. Org. Chem., 1997, 62(20):6974~6977. doi: 10.1021/jo971046m
G Piancatelli, F Leonelli, N Do et al. Org. Synth., 2006, 83:18~23. doi: 10.15227/orgsyn.083.0018
X Q Li, W K Wang, C Zhang. Adv. Synth. Catal., 2009, 351(14-15):2342~2350. doi: 10.1002/adsc.v351:14/15
C Zhang, W K Wang, T He. Synthesis, 2012, 44(19):3006~3014. doi: 10.1055/s-00000084
H Tohma, S Takizawa, T Maegawa et al. Angew. Chem., 2000, 112(7):1362~1364. doi: 10.1002/(ISSN)1521-3757
(a) R M Moriarty, H Hu. Tetrahed. Lett., 1981, 22(29):2747~2750; (b) R M Moriarty, O Prakash, P Karalis et al. Tetrahed. Lett., 1984, 25(42):4745~4748; (c) R M Moriarty, H Hu, S C Gupta. Tetrahed. Lett., 1981, 22(14):1283~1286; (d) R M Moriarty, K C Hou. Tetrahed. Lett., 1984, 25(7):691~694; (e) O Prakash, M P Tanwar, S Goyal et al. Tetrahed. Lett., 1992, 33(43):6519~6522; (f) O Prakash, D Kumar, R K Saini et al. Synth. Commun., 1994, 24(19):2167~2172.
S Hara, T Hatakeyama, S Q Chen et al. J. Fluorine Chem., 1998, 87(2):189~192. doi: 10.1016/S0022-1139(97)00144-9
M Yoshida, K Fujikawa, S Sato et al. Arkivoc, 2003, vi:36~42.
(a) M F Greaney, W B Motherwell. Tetrahed. Lett., 2000, 41(22):4467~4470; (b) W B Motherwell, M F Greaney, J J Edmunds et al. J. Chem. Soc. Perkin Transac., 2002, 24:2816~2826.
J Tao, R Tran, G K Murphy. J. Am. Chem. Soc., 2013, 135(44):16312~16315. doi: 10.1021/ja408678p
T V Moskovkina, V I Vysotskii. Org. Khim., 1991, 27(4):833~836.
H Ibrahim, F Kleinbeck, A Togni. Helv. Chim. Acta, 2004, 87(3):605~610. doi: 10.1002/(ISSN)1522-2675
N A Magnus, L Ducry, V Rolland et al. J. Chem. Soc. Perkin Transac., 1997, 16:2313~2318.
(a) Z Wang, M Kanai, Y Kuninobu. Org. Lett., 2017, 19(9):2398~2401; (b) P Saidhareddy, S Ajay, A K Shaw. Tetrahedron, 2017, 73(30):4407~4417; (c) M Fujita. Tetrahedron, 2017, 58(47):4409~4419; (d) R Dagenais, A J Lauriers, C Y Legault. Synthesis, 2017, 49(13), 2928~2932; (e) X Jiang, C Zheng, L Lei et al. Eur. J. Org. Chem., 2018, 12:1437~1442.
I Tellitu, E Dominguez. Tetrahedron, 2008, 64(10):2465~2470. doi: 10.1016/j.tet.2007.12.045
M Celik, C Alp, B Coskun et al. Tetrahed. Lett., 2006, 47(22):3659~3663. doi: 10.1016/j.tetlet.2006.03.137
W Zhong, J Yang, X Meng et al. J. Org. Chem., 2011, 76(24):9997~10004. doi: 10.1021/jo201752y
M Fujita, Y Ookubo, T Sugimura. Tetrahed. Lett., 2009, 50(12):1298~1300. doi: 10.1016/j.tetlet.2009.01.012
X L Wu, G W Wang. Tetrahedron, 2009, 65(43):8802~8807. doi: 10.1016/j.tet.2009.08.069
A Kirschning, E Kunst, M Ries et al. Arkivoc, 2003, (6):145~163.
A Y Koposov, V V Boyarskikh, V V Zhdankin. Org. Lett., 2004, 6(20):3613~3615. doi: 10.1021/ol0484714
K C Nicolaou, N L Simmons, Y Ying et al. J. Am. Chem. Soc., 2011, 133(21):8134~8137. doi: 10.1021/ja202555m
L Liu, D Zhang-Negrerie, Y Du et al. Org. Lett., 2014, 16(2):436~439. doi: 10.1021/ol403321n
L Rebrovic, G F Koser. J. Org. Chem., 1984, 49(24):4700~4702. doi: 10.1021/jo00198a022
M Hachiya, M Ito, T Matsuo et al. Org. Lett., 2011, 13(10):2666~2669. doi: 10.1021/ol200768b
M A Ciufolini, N A Broun, S Canesi. Synthesis, 2007, 24:3759~3772.
C Sabot, B Commare, M A Duceppe et al. Synlett, 2008, 20:3226~3230.
(a) H Baba, K Moriyama, H Togo. Tetrahed. Lett., 2011, 52(33):4303~4307; (b) Y Zhao, W L Yim, C K Tan et al. Org. Lett., 2011, 13(16):4308~4311.
M Kirihara, S Yokoyama, H Kakuda et al. Tetrahedron, 1998, 54(46):13943~13954. doi: 10.1016/S0040-4020(98)00862-X
(a) H Liu, X Wang, Y Gu. Org. Biomol. Chem., 2011, 9(5):1614~1620; (b) H Liu, Y Xie, Y Gu. Tetrahed. Lett., 2011, 52(33):4324~4326; (c) A Pialat, B Liegault, M Taillefer. Org. Lett., 2013, 15(7):1764~1767.
G W Gribble. Hofmann rearrangement. Name Reactions for Homologations-Part Ⅱ, Wiley:Hoboken, NJ2009:164~199.
M Kalesse, D Landsberg. Synlett, 2010, 7:1104~1106.
M W Justik, G F Koser. Tetrahed. Lett., 2004, 45(32):6159~6163. doi: 10.1016/j.tetlet.2004.06.029
H R Khatri, J Zhu. Chem. Eur. J., 2012, 18(39):12232~12236. doi: 10.1002/chem.201202049
T Abo, M Sawaguchi, H Senboku et al. Molecules, 2005, 10(1):183~189. doi: 10.3390/10010183
Y Sun, X Huang, X Li et al. Adv. Synth. Catal., 2018, 360(6):1082~1087. doi: 10.1002/adsc.v360.6
L F Silva, Jr., R S Vasconcelos, M A Nogueira. Org. Lett., 2008, 10(5):1017~1020.
L Kurti, P Herczegh, J Visy et al. J. Chem. Soc. Perkin Transac, 1999, 4:379~380.
K C Guerard, C Chapelle, M A Giroux et al. Org. Lett., 2009, 11(20):4756~4759. doi: 10.1021/ol902000j
G Jacquemot, S Canesi. J. Org. Chem., 2012, 77(17):7588~7594. doi: 10.1021/jo301408j
H Fujioka, H Komatsu, T Nakamura et al. Chem. Commun., 2010, 46(23):4133~4135. doi: 10.1039/b925687c
K Hata, H Hamamoto, Y Shiozaki et al. Tetrahedron, 2007, 63(19):4052~4060. doi: 10.1016/j.tet.2007.02.118
T Dohi, M Ito, I Itani et al. Org. Lett., 2011, 13(23):6208~6211. doi: 10.1021/ol202632h
H Tohma, M Iwata, T Maegawa et al. Tetrahed. Lett., 2002, 43(50):9241~9244. doi: 10.1016/S0040-4039(02)02150-0
S R Taylor, A T Ung, S G Pyne et al. Tetrahedron, 2007, 63(46):11377~11385. doi: 10.1016/j.tet.2007.08.082
Y Kita, H Watanabe, M Egi et al. J. Chem. Soc. Perkin Transac., 1998, 4:635~636. http://pubs.rsc.org/EN/content/articlehtml/1998/p1/a709099d
Y Kita, M Egi, M Ohtsubo et al. Chem. Commun., 1996, 19:2225~2226.
(a) T Dohi, K Morimoto, N Takenaga et al. J. Org. Chem., 2007, 72(1):109~116; (b) T Dohi, M Ito, N Yamaoka et al. Tetrahedron, 2009, 65(52):10797~10815; (c) T Dohi, K Morimoto, Y Kiyono et al. Org. Lett., 2005, 7(4), 537~540.
T Dohi, K Morimoto, Y Kiyono et al. Chem. Commun., 2005, (23):2930~2932. doi: 10.1039/b503058g
(a) T Dohi, K Morimoto, A Maruyama et al. Org. Lett., 2006, 8(10):2007~2010; (b) N A Waghmode, A H Kalbandhe, P B Thorat et al. Tetrahed. Lett., 2016, 57(6):680~683.
Y Kita, K Morimoto, M Ito et al. J. Am. Chem. Soc., 2009, 131(5):1668~1669. doi: 10.1021/ja808940n
S Rihn, M Erdem, A De Nicola et al. Org. Lett., 2011, 13(8):1916~1919. doi: 10.1021/ol200189y
(a) J Buendia, B Darses, P Dauban. Angew. Chem., 2015, 127(19):5789~5793; (b) H Zhang, Y Su, K H Wang et al. Org. Biomol. Chem., 2017, 15(25):5337~5344.
J A Souto, Y Gonzalez, A Iglesias et al. Chem. Asian J., 2012, 7(5):1103~1111. doi: 10.1002/asia.v7.5
D J Chen, Z C Chen. Tetrahed. Lett., 2000, 41(38):7361~7363. doi: 10.1016/S0040-4039(00)00990-4
A De Mico, R Margarita, A Mariani et al. Chem. Commun., 1997, 13:1237~1238. http://pubs.rsc.org/en/content/articlelanding/1997/CC/a701998j
M Bruno, R Margarita, L Parlanti et al. Tetrahed. Lett., 1998, 39(22):3847~3848. doi: 10.1016/S0040-4039(98)00629-7
M Tingoli, M Tiecco, L Testaferri et al. Synth. Commun., 1998, 28(10):1769~1778. doi: 10.1080/00397919808007007
M Kamlar, J Vesely. Tetrahed-Asym., 2013, 24(5-6):254~259. doi: 10.1016/j.tetasy.2013.02.008
D W Chen, Z C Chen. Synth. Commun., 1995, 25(11):1605~1616. doi: 10.1080/00397919508015845
S Combes, J P Finet. Tetrahedron, 1998, 54(17):4313~4318. doi: 10.1016/S0040-4020(98)00126-4

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



















































 下载:
下载:

