催化一直是化学领域最重要的课题之一,因为催化能够有效地实现能量转换和降低生产成本[1 。传统的负载型金属催化剂已经在工业生产、能源转换等领域得到了广泛应用。然而,大多数负载型金属催化剂存在严重的烧结问题,即在较高的温度下或经历长时间反应后金属原子/颗粒间会发生团聚。造成催化剂活性位点的密度和稳定性的大幅下降,导致实际应用中要经常更换催化剂,无形中提高了催化剂使用成本,降低了工业生产效率[2 3 。
张涛等[4 5 于2011年最早实现了用化学方法制备负载型单原子催化剂,单原子催化的研究兴起至今已经引发了非常广泛的兴趣。作为催化领域的后起之秀,单原子催化剂在保证高催化活性与高选择性的同时,最大限度地提高了金属原子利用率,能够降低生产成本,是工业上最为理想的催化剂之一[6 8 。虽然单原子催化剂拥有传统催化剂不可比拟的优点,但也存在着一些不足,例如当金属粒子尺寸缩小到原子级水平时,比表面积急剧增大,导致金属表面自由能急剧增加[9 11 。随着金属负载量的增加,在制备和反应时易发生团聚形成较大的颗粒,从而导致催化剂失活[12 13 。因此,合成抗烧结的超稳定型单原子催化剂是一个具有挑战性的课题,对单原子催化的发展有着重要意义。
本文首先介绍了近年来抗烧结的超稳定型单原子催化剂的可控合成领域的研究进展;其次,对该类单原子催化剂的催化应用进行了总结;最后展望了该类单原子催化剂的发展前景。
1.
抗烧结单原子催化剂的可控合成
在过去的半个世纪中,单原子催化剂的合成方法飞速发展。在发展初期,比较典型的是以浸渍法和共沉淀法为代表的一类湿化学方法[4 13 。这类方法可以很简便地制备单原子催化剂,不涉及其他复杂的步骤,但利用这种方法合成的单原子催化剂金属负载量往往较低,催化活性往往不能满足实际应用需求[14 。当提高金属负载量时,在金属离子被还原及催化反应的过程中,由于金属原子与载体之间相互作用微弱,易发生团聚烧结[15 16 。面对这些问题,科学家们对耐烧结的超稳定型单原子催化剂的合成方法进行了深入的研究,发展出了多种新型合成方法(见图 1
图 1
1.1
高温热解法
高温热解法是一种通过对负载有金属前驱体的材料进行高温处理,使得金属前驱体分解,并将得到的金属原子通过配位形式锚定在载体缺陷位点上的单原子材料合成方法。对于许多含有N、S、P等含孤对电子的杂原子的材料(如有机聚合物、金属有机骨架(MOFs)等),在热解过程中,其所含有的杂原子对金属原子有很强的配位锚定作用,可以在一定程度上抑制金属原子间的团聚,这使得它们在抗烧结单原子材料的合成中得到了广泛应用,可以在避免金属原子团聚的同时显著提高金属负载量[17 18 。在这一方面,笔者等[19 23 进行了大量研究,利用MOFs和高分子聚合物实现了多种高金属载量的稳定型单原子材料的合成。
1.2
高温迁移法
直接将金属或金属氧化物块体转化为金属单原子具有潜在的工业应用价值。近些年来科学家们发展出了一类高温迁移策略:通过高温激发金属块体释放出可迁移的金属物种,并采用合适的载体对该金属物种进行捕获,可以成功获得单原子材料。这一过程的关键是创造合适的合成条件,将金属原子从块体中释放出来,并在金属单原子和锚定基底之间形成新的化学键。
1.2.1
高温气相迁移
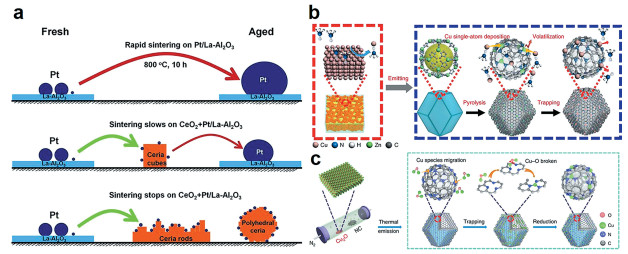
2016年,Jones等[24 利用高温气相迁移在CeO2 载体上获得了Pt单原子。如图 2(a) 2 O3 载体(Pt/La-Al2 O3 )与CeO2 粉末混合均匀,于800℃的氧化氛围中熟化。在这个过程中,Pt纳米颗粒首先以挥发性的PtO2 释放出来,然后迁移的Pt物种会被CeO2 捕获并锚定,形成耐高温的Pt单原子催化剂。他们采用不同形貌的CeO2 进行了实验,结果表明,与立方体CeO2 相比,含有更多缺陷的多面体状和纳米棒状的CeO2 更容易捕获可迁移的Pt物种,并实现Pt的原子级分散。这说明合适的载体对俘获随着气流迁移的金属物种具有非常关键的作用。2018年,笔者等[25 报道了一种简便高效的气相迁移方法,在流动的氨气气氛下通过高温热处理可直接将金属块体转化为负载在N掺杂多孔碳上的金属单原子。将泡沫Cu和ZIF-8粉体分别放置在两个瓷舟中。首先在1173K流动氩气氛下进行热处理,将ZIF-8转化为富含缺陷的N掺杂多孔碳;之后将氩气流转换为氨气流继续进行热处理,这一过程中流动的氨分子通过强Lewis酸碱相互作用与泡沫Cu表面的Cu原子配位形成挥发性的Cu(NH3 )x 3 )x 4 活性位点(图 2(b) 3 )x [26 。如图 2(c) 2 O粉末和N掺杂碳基底分别放入两个瓷舟中。在流动的N2 氛围中进行热处理,这一过程中表面的Cu2 O通过一个热释放过程形成挥发性物质。之后,挥发性Cu物种迁移到N掺杂碳基底的表面被其捕获并锚定,获得Cu单原子催化剂(Cu-ISAS/N-C)。该合成方法可推广到以Mo、Sn金属氧化物为金属源合成Mo、Sn单原子催化剂。
图 2
1.2.2
高温固相迁移
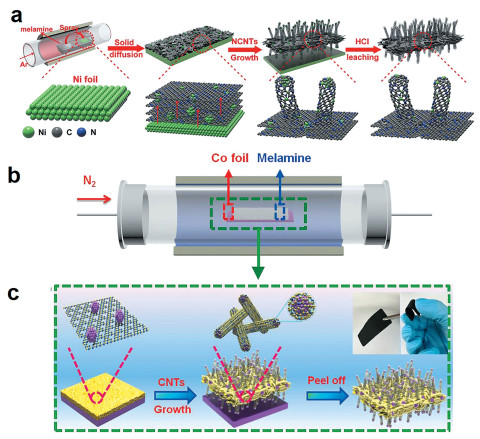
除了高温气相迁移可实现单原子催化剂的大规模制备,我们还发展了一系列高温固相迁移的合成方法。例如,2019年报道了Ni金属块材在N掺杂碳材料上的高温固相扩散作用,能够应用于具有分级自支撑结构的Ni单原子催化剂的合成[27 。如图 3(a) 3 N4 覆盖在Ni箔表面,进而转化为CN层并产生大量不饱和碳空位。在Ni-N配位的强Lewis酸碱相互作用的驱动下,Ni箔表面的Ni原子会扩散到CN层中占据碳空位并形成小的纳米颗粒。与此同时,Ni箔向CN层的定向物质流动会被反向流向Ni箔的C所平衡。在从Ni箔衍生出的Ni“种子”的催化下,一维N掺杂碳纳米管(N-CNT)会垂直于二维CN层表面生长从而形成层次结构。C3 N4 会在持续的高温下气化,为N-CNT的生长提供C、N源。最后通过酸浸步骤溶解CN层中的大部分Ni“种子”,即可得到两面均生长出大量N-CNT的负载Ni单原子的碳纸(H-CPs)。类似地,利用同样的策略由Co金属块材原位固相迁移诱导合成纳米Co封装的三维导电薄膜(Co/CNFs,如图 3(b) (c) [28 。所得材料对电催化氧还原反应(ORR)、析氧反应(OER)和析氢反应(HER)都表现出了出色的催化活性和稳定性。
图 3
综上所述,高温热迁移可以实现直接从多种金属/金属氧化物块体向金属单原子转变,避免了传统合成方法涉及的繁琐步骤,为大规模工业生产应用提供了可能;同时,在高温下合成的单原子催化剂有着杰出的热耐受性和抗烧结性能。
1.3
杂原子掺杂热原子化法
将团聚的金属颗粒转变为单原子是一种新颖的单原子材料合成方式,通过这种方式可以再生已失活的工业催化剂,从而降低催化剂的应用成本。为了实现这一目标,科学家们发展出了一种杂原子掺杂热原子化法:利用高温破坏金属纳米颗粒中的金属-金属键,同时利用金属原子与杂原子之间的强配位相互作用实现热稳定型金属单原子的形成。
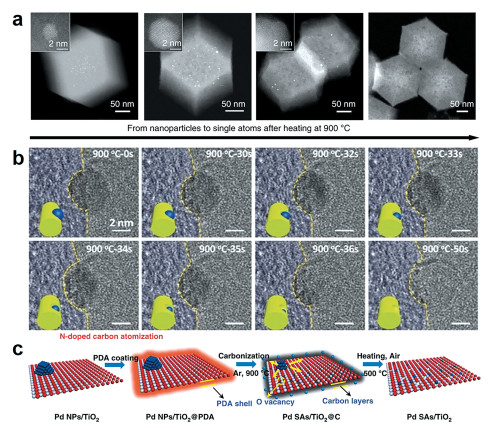
2018年,Li等[29 实现了将贵金属(Pd、Pt、Au)纳米颗粒于900℃以上的惰性气氛中转化为热稳定的金属单原子(图 4(a) [30 深入研究了Ni纳米粒颗粒(Ni NPs)在无定形N掺杂碳材料表面的热原子化过程。在热处理过程中,发现了Ni NPs的一种表面挖掘效应,受到这一效应的影响,表面负载的Ni NPs会刻蚀碳载体表面并钻入下层结构转变为单原子(Ni SAs)。为了进一步阐明表面挖掘效应的内在机理,我们采用原位透射电子显微镜对900℃下Ni纳米颗粒的演变过程进行了分析,如图 4(b) 2 上负载的Pd颗粒转变为Pd单原子(图 4(c) [31 。在传统的煅烧过程中,TiO2 上负载的Pd颗粒会逐渐聚集形成更大的颗粒,然而在利用氮掺杂热原子化法后,出现了完全不同的情况。通过在负载Pd纳米颗粒的TiO2 外包覆一层聚多巴胺后进行热处理,负载的Pd颗粒会受热释放出原子尺度的粒子扩散到CN层,被丰富的N缺陷捕获后形成Pd单原子。将所得到的材料在空气中再次进行热处理后移除CN层,其中Pd单原子会在TiO2 表面被氧缺陷捕获而重新实现原子级分散。这种合成思路为再生已失活的工业催化剂和制备耐烧结的单原子催化剂开拓了一条新的道路。
图 4
除了N以外,其他杂原子, 如S,也可应用于热原子化法合成单原子材料。例如,2018年Zhang等[32 利用S辅助热原子化合成了含硫的原子级钴负载的空心碳(HC)复合材料(S@Con [33 。以CdS纳米棒作为前驱体,通过合理设计阳离子交换反应(CER)中的沉淀溶解平衡,可以控制Cu、Pt、Pd等客体金属的阳离子在CdS纳米棒表面进行原子级/纳米级交换。反应完成后在其外包覆一层含N高分子聚合物进行热解,由于不同金属的沸点不同,低沸点的母体金属(Cd)在高温下会升华,这一过程中CdS中的部分S会与CN基底间发生反应从而实现S掺杂。通过合理的热解温度设计,富含S和N缺陷的CN基底可以及时锚定高沸点的客体金属,从而获得S、N共修饰的单原子材料。
杂原子掺杂热原子化法作为一类将金属纳米颗粒转化为热稳定、耐烧结的金属单原子的方法,展现出了广阔的催化应用前景。
通过以上方法可以高效可控地合成抗烧结的超稳定型单原子催化剂,但是这些方法依然存在着一些不足。例如,在制备过程中往往需要进行高温处理,从而导致较高的能耗,间接提高了催化剂的生产成本;此外,对于部分通过高温热解与杂原子掺杂热原子化法制备的单原子催化剂,较多的活性位点深入材料内部,不能很好地与反应物接触,从而造成了金属原子利用率的损失。这些是抗烧结单原子催化剂发展过程中必须要解决的问题。
2.
抗烧结的超稳定型单原子催化剂的应用
开发具有高活性、高稳定性、高选择性的催化剂一直是工业催化的最高追求。抗烧结的超稳定型单原子催化剂由于具有独特的结构和充分暴露的活性位点以及高稳定性,在甲烷氧化、选择性加氢、酶催化反应、电化学催化反应等重要领域中展现出了广阔的应用前景[34 。
2.1
甲烷氧化中的应用
由于储量丰富且价格低廉,甲烷已经成为生产基本化学品最有前途的基础材料之一。对于甲烷氧化反应,传统催化剂,如氧化物负载贵金属Pd、Pt、Au、Ru、Rh等的催化剂,通常需要在高温(100~500 ℃)或强酸介质条件下才能破坏高度稳定的C-H键(104kcal/mol),因此,在温和的条件下利用非贵金属催化剂实现甲烷氧化是一个巨大的挑战[35 。
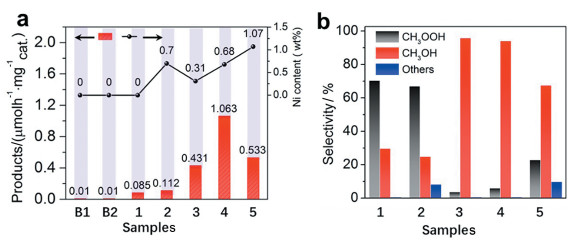
2018年,Cui等[36 报道了一种石墨烯锚定的Fe单原子材料(FeN4 /GN)可以在室温下直接将CH4 转化为C1氧化产物。与以往报道的材料相比,FeN4 /GN催化剂能在较温和的条件下促进CH4 转化为C1氧化产物,同时拥有较低的CO2 的选择性。在反应过程中CH4 首先被氧化成CH3 OH和CH3 OOH,经过一段时间之后生成的CH3 OH会被进一步被氧化成HOCH2 OOH和HCOOH。2019年,笔者等[30 报道了一种通过氮掺杂热原子化法合成的Ni单原子催化剂(CNT@PNC/Ni SAs),在以过氧化氢为氧化剂的条件下,可催化CH4 在50℃的低温下氧化生成CH3 OH。图 5(a) 4 的转化速率从0.431μmol·h-1 ·mgcat -1 提高至1.063μmol·h-1 ·mgcat -1 。对于催化剂的选择性,在10h的CH4 氧化反应之后,CNT@NC的催化产物以CH3 OOH为主,CH3 OH仅占20%~30%(图 5(b) 3 OH的比例高达94.2%。DFT计算表明,Ni单原子位点在CH4 活化过程中起主导作用,热原子化过程中的表面挖掘效应会诱导Ni-N4 中心附近产生碳缺陷,而这些缺陷会影响Ni-N4 活性位点的电子云密度分布,从而改变活性位点与反应中间体的键合强度,大大提高催化活性。
图 5
2.2
氢化反应中的应用
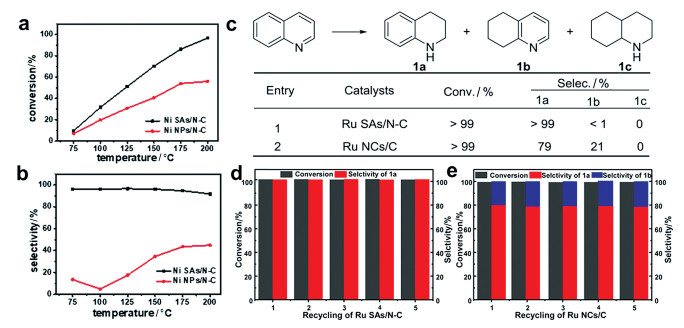
氢化反应是有机化合物的工业生产过程中非常重要的反应,常见的有烯烃、炔烃等不饱和键的加氢反应。例如,聚乙烯生产中使用的乙烯是由石脑油热裂解产生的,通常含有微量的乙炔。然而少量的乙炔会使催化剂中毒,降低生产的聚乙烯的质量。因此获得高效稳定的催化剂以从乙烯气流中去除混有的少量乙炔显得尤为重要[37 38 。2017年,我们利用高温热解合成了Ni单原子锚定在N掺杂多孔碳中的材料(Ni SAs/N-C),对乙炔加氢的催化性能显示出了替代Pd基催化剂的巨大潜力(图 6(a) (b) [39 。在模拟工业烯烃馏分净化的测试条件下,Ni SAs/N-C对乙炔的转化率和对产物乙烯的选择性均超过90%,而Ni NPs/N-C对乙烯的选择性在所有测试温度下都小于55%。尽管Ni SAs/N-C和Pd-Ag合金催化剂的转化率都接近100%,但Ni SAs/N-C高达90%的选择性超过了Pd-Ag合金催化剂。Ni SAs/N-C催化剂具有专门用于乙炔吸附和乙烯解吸附的Ni-Nx
图 6
抗烧结单原子材料不仅能够应用于工业乙烯中乙炔的选择性氢化,还可以催化其他氢化反应。例如,笔者等[40 以UiO-66-NH2 为载体通过高温热解法获得了一种Ru单原子催化剂,其对功能化喹啉类化合物的选择性加氢有着良好的催化性能。如图 6(c) 图 6(c) 1a 图 6(d) (e)
2.3
其他应用
除了上述热催化反应,通过前文所述方法制备的抗烧结单原子催化剂在多个领域都有着广阔的应用前景。例如在酶催化领域,笔者等于2019年通过高温气相迁移法得到了一种活性位点结构类似于天然含血红素酶的Fe单原子催化剂,有着优异的过氧化物酶、氧化酶和过氧化氢酶的类酶活性,比Fe3 O4 纳米酶的活性高出40倍[41 。2020年,我们通过模板辅助的高温热解法获得了一种N掺杂多孔碳负载的Fe单原子位点纳米酶,具有优良的类过氧化物酶活性,可应用于人体血糖检测[42 。在电催化领域,抗烧结的超稳定型单原子催化剂对电催化ORR、OER、HER和二氧化碳还原反应(CO2 RR)等关键能量转换反应的催化应用多有报道。例如,Li等[43 利用高温热解法获得了一种具有孤立Co单原子位点的中空N掺杂碳球材料(ISAS-Co/HNCS),有着良好的酸性ORR催化性能,其半波电位(E 1/2 )接近于商业Pt/C。2019年,笔者等[27 利用N掺杂碳材料和Ni金属块体之间的高温固相迁移合成了分层、自支撑的Ni单原子催化剂(H-CPs),可直接用作无粘接剂的电极实现高效率CO2 RR,在-1.0V(相对于RHE)的电势下有着高达48.44mA·cm-2 的电流密度。其对CO产物的法拉第效率高达97%,且对CO产物的选择性在-0.7V到-1.2V(相对RHE)的电势范围内可以保持在90%以上。2019年,我们通过高温热解法得到了一种负载Fe单原子的多孔N掺杂碳材料(Fe SAs/N-C),其金属负载量达3.5(wt)%[44 。Fe SAs/N-C对酸性与碱性ORR都表现出了卓越的活性与稳定性。
3.
总结与展望
本文对抗烧结的超稳定型单原子催化剂的可控合成及催化应用的研究进展进行了总结。烧结现象一直是工业催化剂应用道路上的一块绊脚石,为了实现抗烧结单原子材料的可控合成,科学家们研究并发展了一系列新型合成方法,例如高温热解法、高温迁移法、杂原子掺杂热原子化法等。这些方法的发展为单原子催化剂的工业化生产铺垫了道路。在催化应用方面,抗烧结单原子催化剂由于其超高的稳定性,在热催化领域应用广泛。除此之外,科学家们还尝试了将该类单原子催化剂应用于酶催化反应、电催化反应以及其他催化反应,至今已经取得了很多突破。
单原子催化剂可以实现原子利用率最大化,这为金属资源的合理利用和促进催化系统的低成本发展带来了希望。充分暴露的活性位点、独特的电子结构和增强的金属-基底间的相互作用使单原子催化剂具有卓越的催化性能和稳定性。这些优秀的性质推动了其在各种工业催化反应中的应用。虽然抗烧结单原子催化剂的发展已经取得了一些成就,但在将来的发展中依然需要注意以下几个问题:(1)现阶段对实现金属活性位点的精确调控依然存在着挑战,需要通过配位结构设计发展出更加精确可控的合成方法;(2)工业催化中催化剂的需求量往往较高,目前的合成方法还处于实验阶段,距离大规模的工业生产应用还有一定距离。随着单原子催化剂的制备方法与表征手段逐渐丰富,其应用领域也日益拓宽。相信抗烧结的超稳定型单原子催化剂在能源、环境、生物医学等领域将会有大量重要的研究成果涌现。

 下载:
下载:

 下载:
下载:







