表 1
实验条件
Table 1.
Reaction test conditions

汞是一种危害性极大的重金属,具有剧毒性、持久性和生物富集性,近年来其排放受到了极大的关注[1, 2]。应用于脱硝系统中的SCR催化剂被证明同时拥有将Hg0氧化为Hg2+的能力,因此利用SCR催化剂将难溶于水的Hg0氧化为易溶于水的Hg2+,并利用后续的湿法脱硫(WFGD)设备协同脱除Hg2+是一种经济且高效的控制汞排放的方法[3, 4]。
在众多SCR催化剂中,MnOx基催化剂具有高效、低廉及低温适应性等特点,受到了广泛研究[5, 6]。但是纯MnOx的比表面积小、结晶度高,使其并不具备如此高的催化性能[7],将其他金属掺杂到MnOx晶体结构中是改善其催化性能的有效方法。在MnOx的晶体结构中添加钾离子,形成了由2×2[MnO6]八面体组成的一维孔结构,其中,K+位于一维孔通道中[8-10],即氧化锰八面体分子筛(OMS-2),极大地提高了其比表面积,约为MnOx的10倍[8],并且拥有更丰富的孔结构[11, 12]。对于OMS-2,Mn平均价约为3.8[13],这意味着OMS-2中的大多数Mn以Mn4+的形式存在,其被认为比Mn3+对Hg0的氧化更有利[14]。虽然已有研究证明OMS-2在Hg0氧化方面具有优良的性能[15],但是与大多数SCR催化剂类似,OMS-2同样存在抗硫(SO2)性差的缺陷[14]。
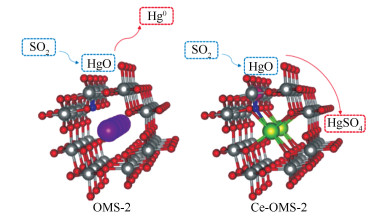
截至目前,SO2抑制SCR催化剂上Hg0的氧化机制一般认为有SO2与HCl竞争吸附[16, 17];催化剂中毒形成惰性硫酸盐物质[18-20]。此外,前期研究发现,在OMS-2催化剂上甚至可引起已被氧化的HgO再次被SO2还原为Hg0 [15]。因此,深入研究SO2还原HgO的反应机理,开发出具备优良抗硫性能的催化剂是当前亟待解决的科学问题。铈(CeO2)基催化剂上存在较多的Ce3+/Ce4+氧化还原对,可在氧化还原条件下通过价态变化(CeO2(Ce4+)←Ce2O3(Ce3+))产生丰富不稳定的氧空穴,储存更多的表面晶格氧,表现出强大的储氧能力,极大程度提高汞的氧化能力[21]。此外,Ce的亲硫性较Mn更强,将Ce掺杂进Mn基催化剂中,SO2会先与Ce反应,从而保护活性组分Mn,减轻其受SO2的侵害[22],有望弥补OMS-2催化剂抗硫性差的缺陷使其具有更广阔的应用前景。
因此,本研究旨在利用Ce改性OMS-2以增强催化剂的抗硫性能,并降低其表面HgO被还原的程度,进一步获得具备更高性能协同脱硝脱汞的SCR催化剂。为此,本研究采用无溶剂法、水热法分别制备了OMS-2及Ce改性的OMS-2(Ce-OMS-2),利用固定床实验对比考察了SO2在两种催化剂表面对Hg0氧化以及HgO还原的性能,综合热力学计算结果探讨了SO2与HgO发生还原作用时的反应机理,以期为燃煤电厂制定高效经济的Hg0排放控制策略提供参考。
采用无溶剂法制备OMS-2催化剂。首先将22.05 g Mn(Ac)2·4H2O与9.48 g KMnO4(物质的量比3:2)在研钵中研磨混合均匀,然后放入80 ℃干燥箱中,加热4 h,得到黑色黏性产物。将该黑色产物用去离子水反复清洗直至滤液澄清为止,再次放入80 ℃干燥箱中干燥12 h。最后,将干燥所得固体研磨成60-80目粒径的颗粒。
为了将Ce能够更好地以离子形态掺杂进入OMS-2催化剂,采用水热法制备Ce-OMS-2催化剂。将4.3422 g Ce(NO3)3·6H2O溶解于40 mL去离子水中,将4.7412 g KMnO4加入上述溶液并不断搅拌直至完全溶解。之后将溶液转移至100 mL的特氟龙瓶中,并密封于钢制反应釜中,于120 ℃下反应24 h。待反应釜取出冷却至室温后,将反应所得沉淀用去离子水反复清洗直至滤液澄清为止,再将其放入干燥箱中干燥。最后,将干燥所得固体研磨成60-80目粒径的颗粒。
固定床实验系统主要由配气系统、反应器、汞测试系统以及烟气测试系统组成。常规烟气包括高纯N2,高纯O2,标准二氧化硫SO2由钢瓶供给,通过D07-19B型质量流量控制器控制各组分的含量。汞渗透管(VICI Metronics)作为汞发生源,置于恒温水浴锅中,以高纯N2作为载气引入混合气体中,通过调节恒温水浴锅的温度向模拟烟气中提供稳定含量的Hg0,气体总流量1 L/min。SCR催化剂填充在内径为10 mm高硼硅玻璃反应器中,并通过管式炉控制温度。采用VM 3000测汞仪在线监测进口汞含量(Hgin0)和出口汞含量(Hgout0)。Hg0氧化效率(xoxi)采用公式(1)计算:
|
$ x_{\mathrm{oxi}}(\%)=\frac{\mathit{\Delta} \mathrm{Hg}^{0}}{\mathrm{Hg}_{\mathrm{in}}^{0}}=\frac{\mathit{\Delta} \mathrm{Hg}_{\mathrm{in}}^{0}-\mathit{\Delta} \mathrm{Hg}_{\mathrm{out}}^{0}}{\mathit{\Delta} \mathrm{Hg}_{\mathrm{in}}^{0}} \times 100 \% $ |
(1) |
本模块共设计六组实验,实验条件见表 1。
 下载:
导出CSV
下载:
导出CSV
| Test | Catalyst | Carrier gas | Temp. t/℃ | Flow /(L·min-1) |
| 1 | OMS-2 | N2,N2+0.05%SO2,N2+0.1%SO2,N2+0.1%SO2+4%O2 | 150 | 1 |
| 2 | Ce-OMS-2 | N2,N2+0.05%SO2,N2+0.1%SO2,N2+0.1%SO2+4%O2 | 150 | 1 |
| 3 | Ce-OMS-2#1 | N2 | 100-700 | 0.5 |
| Ce-OMS-2#2 | ||||
| 4 | OMS-2* | N2+0.05%SO2 | 150 | 1 |
| 5 | Ce-OMS-2* | N2+0.05%SO2 | 150 | 1 |
| 6 | 10 mg HgO | N2+0.05%SO2 | 150 | 1 |
| note:“#1”, the catalyst was pretreated under N2+4%O2+Hg0 for 1 h; “#2”, the catalyst was pretreated under N2+4%O2+0.05%SO2+Hg0 for 1 h; “*”, the catalyst pretreated under N2+4%O2+500 μg/m3 Hg0 for 7 d | ||||
第1组与第2组实验分别测试了OMS-2及Ce-OMS-2催化剂在有/无O2及不同SO2浓度条件下的汞氧化效率。为探究相比于OMS-2催化剂,Ce-OMS-2催化剂表现出更优的汞氧化性能及抗硫性能的机理,设计了第3组Hg0-程序升温脱附实验,以分析在SO2存在时,Ce-OMS-2催化剂表面的汞物种。第4组及第5组实验通过预处理的方式使催化剂表面负载HgO,之后通过通入SO2气氛,检测催化剂下游是否有Hg0释放出,并对比两种催化剂表面HgO被还原的情况。第6组实验采用纯HgO试剂与SO2反应,以验证该反应是否为自发反应。实验中催化剂的用量为10 mg。
本研究对两种催化剂分别进行了包括比表面积(BET)、X射线衍射分析(XRD)、X射线电子能谱分析(XPS)、电感耦合等离子体/光发射光谱分析(ICP)表征,并采用傅里叶红外光谱分析了经过SO2预处理和未预处理时OMS-2催化剂上汞的形态分布。
综合运用HSC Chemistry[23]软件9.3版中的反应模块(Reaction Equation)和平衡相组成模块(Equilibrium Composition),对HgO在SO2气氛下的可能性反应进行模拟分析。具体模拟计算过程如下:首先根据实验数据和现象在Equilibrium Composition模块输入已明确的反应物HgO和SO2,然后根据质量守恒定律初步猜测可能的反应产物,设置反应压力和温度,输入目标函数(即HgO=0)。最后根据计算结果分析产物和反应物的关系,保留与反应物有关联的产物并剔除那些与反应物没有关联的产物,重复校对筛选多次,直到目标函数可以实现且输入界面的初始反应物与产物之间有密切的关联性,初步得到反应物与产物之间的化学反应途径,随后将反应物与产物在Reaction Equation模块进行配平并计算化学反应参数,观察计算结果是否符合吉布斯自由能判据,如若符合,则进行反应机理的分析,如若不符合,则更换产物继续计算,直到出现可以满足条件的产物。
图 1为150 ℃下SO2对催化剂上Hg0氧化性能的影响。从图 1中可以看出,在极高的空塔速度5400000 h-1(明显超燃煤电厂中烟气的空塔速度),纯N2气氛下,OMS-2催化剂的Hg0氧化效率为41.5%,掺杂Ce后,Hg0氧化效率提高到了54.5%。而添加4%的O2后,OMS-2催化剂上Hg0氧化效率达56%,Ce-OMS-2催化剂上Hg0氧化效率更是高达89%,对比纯N2气氛下,两者的Hg0氧化效率均得到大幅度提高。这是因为反应遵从Mars-Maessen机理,气相O2吸附到催化剂表面不断地补充被Hg0消耗的表面活性氧,使汞的催化氧化反应朝正向持续进行[24-26]。当往N2中依次添加0.05%、0.1% SO2后,OMS-2催化剂的汞转化效率分别急剧降低至14.6%和11.5%,氧化性能得到了极大的抑制。然而,与之形成鲜明对比的是,在添加0.05%、0.1%SO2后,Ce-OMS-2催化剂的汞转化效率不但没有降低,反而分别提升至84%、83.6%,甚至在有氧条件下可达到90.8%。该组实验结果说明,SO2对于没有掺杂Ce基的OMS-2催化剂会产生强烈的抑制作用,而Ce的改性几乎可以弥补该催化剂抗硫性差的缺陷使其具有更广阔的应用前景。
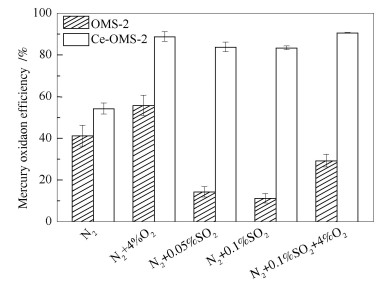
根据比表面积表征结果和表面原子浓度分析(见表 2)可知,OMS-2催化剂的比表面积为173 m2/g,改性后的Ce-OMS-2比表面积为390 m2/g,因此,Ce基掺杂后可以极大地提高OMS-2催化剂的比表面积以及丰富空隙结构。根据ICP的表征结果可以发现,OMS-2中K/Mn的原子比为0.10,接近于分子式K4Mn32O64中的比例,在掺杂Ce后,这一原子比下降到了0.038,Ce/Mn的原子比为0.35,接近于反应物KMnO4与Ce(NO3)3的物质的量比3.0。说明Ce4+成功取代了OMS-2通道结构中的K+,相比于Ce4+取代OMS-2骨架结构上Mn4+,该改性方式能够使催化剂的晶格氧活性更高[27]。因此,Ce掺杂后,OMS-2催化剂在N2和O2气氛下Hg0的氧化效率显著提高,特别是在4%O2气氛下,氧化效率可以达到90.8%。
下载:
导出CSV
| Catalyst | Surface area A/(m2·g-1) | Surface atom concentration | ||||||
| by ICP | by XPS | |||||||
| K/Mn | Ce/Mn | Mn3+/Mn4+ | Ce3+/Ce4+ | Oα | Oβ | |||
| OMS-2 | 173 | 0.10 | - | 1.07 | - | 79.6% | 20.4% | |
| Ce-OMS-2 | 390 | 0.038 | 0.35 | 1.10 | 0.24 | 68.2% | 31.8% | |
| note: Oα: lattice oxygen; Oβ: surface adsorbed oxygen | ||||||||
OMS-2催化剂的XRD谱图验证了其为锰氧八面体分子筛的隐锰钾矿结构(JCPDS-29-1020)[27]。如图 2(a)所示,在掺杂Ce后,属于OMS-2的特征峰消失了,同时,也未检测到MnOx或者CeO2所对应的特征峰,说明Ce成功地进入了OMS-2结构内部,与ICP表征结果一致。
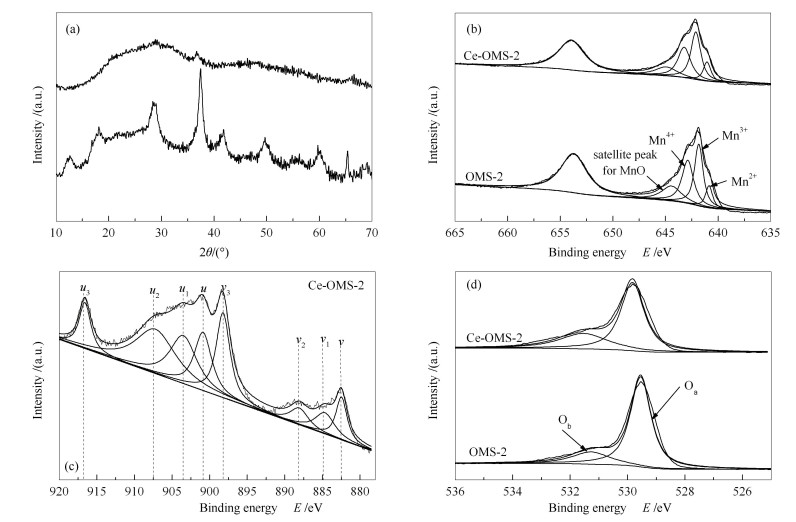
(a)XRD,(b)Mn 2p XPS能谱图,(c)Ce 3d XPS能谱图,(d)O 1s XPS能谱图
(a): XRD patterns; (b): Mn 2p XPS spectra; (c): Ce 3d XPS spectra; (d): O 1s XPS spectra
图 2(b)为Mn 2p XPS能谱图。由图 2(b)可知,无论是OMS-2还是Ce-OMS-2催化剂,Mn都主要以Mn3+、Mn4+存在[15]。且Mn3+/Mn4+的比率分别为1.07和1.10(见表 2),两者几乎没有差别。结合ICP的分析结果发现,Ce-OMS-2催化剂中高价态的Ce4+取代了OMS-2催化剂原本存在的K+后,Mn3+/Mn4+比率并没有增加,据此可推断Ce-OMS-2催化剂存在Mn缺陷,从而使得催化剂能够保持电中性[27]。从这个角度也再一次说明了Ce的改性可以使催化剂的晶格氧活性更高[27],使其在0.05%SO2+N2甚至0.1%高浓度SO2的气氛下也能保持很高的汞氧化能力。
图 2(c)为Ce 3d XPS能谱图,图 2(c)中u3、u2、u(3d3/2)、v3、v2、v(3d5/2)六个特征峰归属于Ce4+,u1、v1特征峰归属于Ce3+[28]。表 2中的结果显示Ce3+ /Ce4+的比率在0.24左右,说明Ce-OMS-2存在丰富的Ce3+ /Ce4+氧化还原对且其主要形态为Ce4+。在氧化Hg0的过程中,铈会在Ce4+
图 2(d)为O 1s XPS能谱图。结合表 2结果可以发现,Ce-OMS-2催化剂中表面吸附氧占总氧(Oα+Oβ)[15]的比例为31.8%,明显高于OMS-2催化剂的20.4%,而表面吸附氧越多说明电子迁移率越高,其氧化性能更好[29]。因此,在添加4%的O2后,OMS-2催化剂的汞转化效率仅为56%,而Ce-OMS-2催化剂的汞转化效率更是高达89%。气相O2吸附到催化剂表面不断补充被Hg0消耗的表面吸附氧,使汞的催化氧化反应朝正向持续进行。
综合所有表征结果可知,Ce基能够极大地改善OMS-2氧化Hg0的原因主要是:获得更大的比表面积及丰富的空隙结构;形成Mn缺陷、提高电子迁移率获得更高的表面吸附氧占比。进而使其在恶劣的SO2气氛下,也能依旧保持优异的汞氧化能力。
关于SO2抑制Hg0的氧化主要有两种观点:第一,催化剂毒化机理,SO2在有氧条件下被氧化为SO3继而与NH3和H2O反应生成(NH4)2SO4等硫酸盐,沉积在催化剂表面导致其失活[30, 31];催化剂的活性组分被SO2硫酸化导致催化剂活性组分减少降低氧化能力,这种观点主要基于催化剂上Hg0的氧化过程。第二,SO2作为烟气中的还原性气氛,可引起已被氧化的Hg0重新转化为Hg0,严重降低SCR催化剂上Hg0的氧化效率[32, 33]。这也很可能是部分电厂SCR装置下游烟气中Hg0比例仍然很高的重要原因[34]。接下来本研究分别对这两种机理进行机理分析。
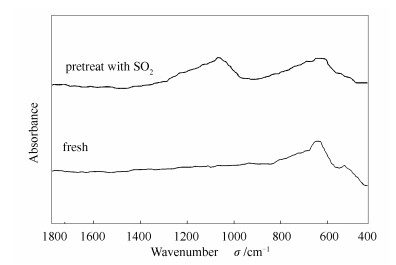
在图 1中,当往N2中添加0.05%、0.1%SO2后,OMS-2催化剂的汞转化效率急剧降低至14.6%、11.5%,氧化性能得到了极大的抑制。将OMS-2催化剂在SO2预处理一段时间后进行FT-IR表征,结果如图 3所示。
由图 3可知,红外谱图中检测到了SO42-的吸收峰[15],而在新鲜催化剂表面并未检测到,说明SO2会与OMS-2反应生成硫酸盐,导致催化剂中毒、表面的活性位点失活从而降低Hg0的转化效率。添加4%O2后,SO2的抑制作用得到一定的缓解,但是汞的氧化效率依旧只有29.5%,低于纯N2气氛下汞的氧化效率,因此抗硫性差会成为OMS-2催化剂应用的限制性因素。
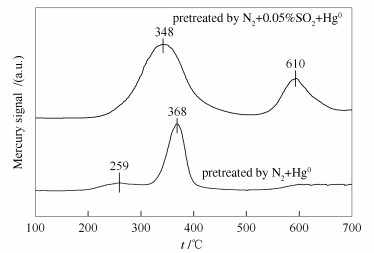
然而,Ce-OMS-2催化剂上汞的氧化效率却没有受到SO2的显著影响。为了探究SO2对Ce-OMS-2催化剂上汞氧化产生的影响机理,开展了Hg0-程序升温脱附实验。Ce-OMS-2催化剂在N2+Hg0的气氛中预处理1 h后,在500 mL/min的N2气流中以10 ℃/min的升温速率从室温升至700 ℃进行汞的脱附,结果见图 4。
由图 4可知,汞的脱附曲线中出现了两个峰值,在259 ℃处出现的平缓的峰归属于催化剂表面的化学吸附Hg0的脱附,在368 ℃出现的峰归属于HgO的分解,可以看出催化剂表面的主要汞物种为HgO[22]。而将Ce-OMS-2催化剂在N2+0.05%SO2+Hg0的气氛中预处理1 h后,HgO的分解峰由368 ℃转移到了348 ℃,且峰变得更宽,说明此时催化剂表面吸附了更多的HgO,同时在610 ℃处出现了新峰,其归属于HgSO4的分解[35, 36]。说明了SO2可在Ce-OMS-2催化剂表面将Hg0氧化为HgSO4,减少催化剂的硫酸化中毒现象,从而提高催化剂上Hg0的氧化效率。
SO2引起催化剂发生硫酸盐中毒现象是SO2抑制SCR催化剂上Hg0氧化的常见机理之一,但是SO2对OMS-2催化剂上Hg0的氧化起到了极其严重的影响,SO2本身是一种常见的还原性气体,而HgO具有一定的氧化性,是否存在其他的抑制机制协同作用,例如Hg2+再次还原为Hg+甚至Hg0导致Hg0氧化效率骤减是我们接下来考察的重点。
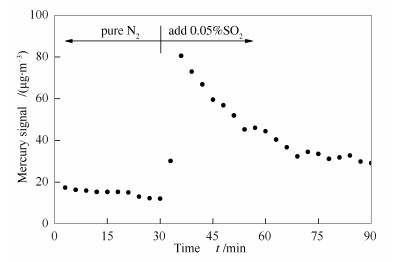
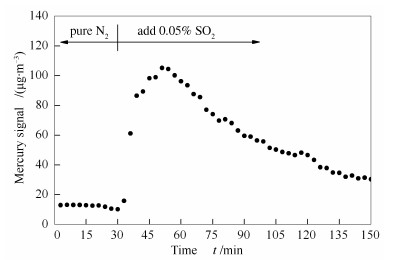
本研究通过第4-6组实验对这一猜想进行了验证。如图 5所示,将OMS-2在N2+4%O2+Hg0的气氛中预处理7 d使其表面负载HgO,并用N2冲洗脱附表面吸附的Hg0直至出口处Hg0趋近于0且不再变化,接着在150 ℃下,通入0.05%SO2(N2作平衡气),观察到出口处析出大量Hg0。该实验说明已被OMS-2氧化得到的HgO在经过SO2后又再次被还原成了Hg0,从而明显降低了OMS-2催化剂上Hg0的氧化效率,与图 1中实验结果取得一致性。
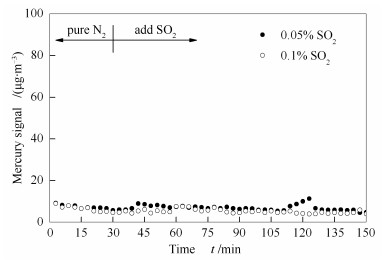
与OMS-2催化剂表面发生HgO还原现象不同的是,当0.05%SO2通过负载了HgO的Ce-OMS-2催化剂时,在反应器出口并没有明显观察到Hg0的增加,甚至增加SO2的体积分数到0.1%,仍未见出口处Hg0含量升高,结果见图 6。结合前述表征结果可分析,引起该现象的原因可能是:Ce改性得到的催化剂比表面积大,可以在催化剂表面通过化学吸附吸附更多的Hg0;表面吸附氧(Oβ)占比高,可提供的活性位点多,导致Ce-OMS-2催化剂表面尽管真的存在一定量HgO被SO2还原为单质汞,也会被迅速重新氧化成HgO,提高了表观Hg0氧化效率。因此,相对于OMS-2催化剂,Ce-OMS-2催化剂不仅可以提高其在N2和O2气氛下的Hg0氧化效率,也可以显著提高其抗硫性差的缺陷。
为了进一步验证该猜想,设计实验将纯HgO试剂与SO2进行直接反应,结果如图 7所示,150 ℃下,当纯HgO中仅通入N2时,反应器出口观察到微量的Hg0,并且成递减趋势,当向反应体系中添加0.05%SO2后,反应器出口Hg0浓度急剧增加,说明HgO可与SO2发生自发反应,直接将其转化为Hg0释放到反应器出口。
SCR催化剂上Hg0的氧化过程与SO2的氧化过程类似,Hg0的氧化是笔者所期望的,但是笔者必须控制SO2的氧化在较低的水平,因为SO2氧化生成的三氧化硫(SO3)可以引起设备腐蚀以及催化剂堵塞等严重问题[20]。然而,目前,涉及SCR催化剂上Hg0氧化与SO2氧化两个相似过程之间的相互作用,SO2氧化过程对Hg2+还原过程的影响的相关报道极少。如果忽略该过程的反应机理而任由SO2的氧化过程增强HgO的还原过程,将会严重降低SCR催化剂上Hg0的氧化效率,加重燃煤电厂下游烟气中Hg0的比例。
本研究采用热力学平衡组分模块对OMS-2催化剂上HgO被重新还原为Hg0的现象进行了分析。OMS-2是作为反应的催化剂,计算时其前后物质不变,因此,反应物为SO2和HgO。对于反应产物,实验现象观察到了Hg0再次释放,因此,Hg0是反应产物之一。其次,由于温度影响可能引起HgO的分解产生O2,以及SO2被O2氧化产生SO3,所以产物理应含有O2和SO3。最后,适当增加部分反应中间产物如HgSO4、HgSO3甚至HgS等不同价态的汞盐。初步计算结果显示HgSO3和HgS在反应产物中存在极少,可以忽略不计。因此,首先定下所有反应产物包括Hg0、O2、SO3和HgSO4,随后将所有反应物和产物输入到Equilibrium Composition模块,各反应物及产物的物质的量见表 3,得到的计算结果见图 8。
下载:
导出CSV
| Species | HgO | SO2 | O2 | SO3 | Hg | HgSO4 |
| Amount of substance /kmol | 1 | 1 | 0 | 0 | 0 | 0 |
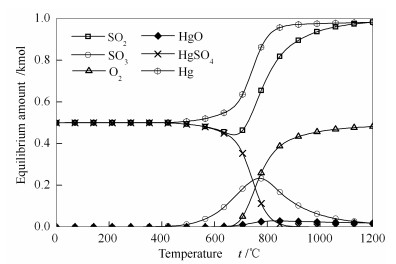
当温度为0-350 ℃,反应体系中SO2已由初始的1 kmol转变为了0.5 kmol,而HgO减少为0,且该过程有Hg0和HgSO4的生成,根据质量守恒定律,可得到该阶段的反应方程式如式(2)所示。
|
$ \mathrm{SO}_{2}(\mathrm{~g})+2 \mathrm{HgO}(\mathrm{g})=\mathrm{Hg}(\mathrm{g})+\mathrm{HgSO}_{4} $ |
(2) |
当温度为500-1000 ℃,此时SO2含量继续减少,并开始有大量的SO3生成,鉴于前述步骤中HgO已经完全消耗,这里与SO2作用生成SO3的只能是HgSO4。与此对应的是,在该温度区间,前一阶段生成的HgSO4在急剧减少,可得到该部分的反应机理如式(3)所示。
|
$ \mathrm{SO}_{2}(\mathrm{~g})+\mathrm{HgSO}_{4}(\mathrm{~g})=\mathrm{Hg}(\mathrm{g})+2 \mathrm{SO}_{3}(\mathrm{~g}) $ |
(3) |
此外,在该区间还开始有大量的氧气生成,这是SO3(g)分解得到O2(g)和SO2(g)的结果,同时也能充分解释该区间SO2(g)虽有减少但是很快又增加的原因。当温度高于1000 ℃时,SO3(g)进一步分解此时各反应逐渐趋向最稳定的状态,最终产物只剩Hg0、O2和SO2。
通过该计算结果,可以直观了解到,OMS-2催化剂上可能发生的HgO经SO2作用的还原途径,且该计算结果也可验证图 4中Ce-OMS-2催化剂上610 ℃峰是由于SO2最初氧化Hg0得到的HgSO4被SO2再次还原析出造成的,即在SO2浓度较低或者温度较低的过程中,可能存在被SO2还原HgSO4的反应。所以在第4组实验中观察到Hg0先被部分氧化后迅速大量释放的机理可解释为SO2在第一阶段参与了Hg0的氧化,并生成SO3和HgSO4。但随着反应的进行,SO2再次将HgSO4还原为单质汞,导致出口处检测到大量的Hg0。但是SO2引起Hg0氧化效率降低的过程不一定必须要经过这两个过程,如第6组实验结果所示,当纯HgO经过SO2气氛时,也可观察到出口处有大量的Hg0释放,说明在没有催化剂存在的条件下,SO2也可以直接与HgO发生氧化还原反应,生成大量的Hg0。这一机理可体现为反应方程式(2)和(3)的综合,即HgSO4只是作为一个中间产物出现在了SO2抑制Hg0氧化的反应过程中,该直接还原的反应方程式如(4)所示,反应机理见图 9。
|
$ \mathrm{SO}_{2}(\mathrm{~g})+\mathrm{HgO}(\mathrm{g})=\mathrm{Hg}(\mathrm{g})+\mathrm{SO}_{3}(\mathrm{~g}) $ |
(4) |
OMS-2催化剂的抗硫性较差。相比于N2气氛,反应体系中添加0.05%和0.1%SO2后,OMS-2催化剂的汞转化效率急剧降低至14.6%、11.5%,氧化性能得到了极大的抑制。究其原因主要是因为SO2会使OMS-2催化剂发生中毒现象,甚至会引起其表面的HgO被再次还原成Hg0,使得表观汞氧化效率降低。
采用Ce改性OMS-2催化剂后,催化剂的比表面积由179 m2/g提高至了390 m2/g,Ce4+成功取代OMS-2八面体孔通道结构中的K+,并形成Mn缺陷,且Ce本身优异的储氧能力,使得Ce-OMS-2表面拥有丰富的表面活性氧,从而保障改性后的催化剂不仅具备更高的汞催化氧化性能,且具备了良好的抗硫性。
SO2还原HgO的过程可自发发生,且可通过多种途径将HgO还原为Hg0。在较低含量SO2和较低温度下生成HgSO4,较高浓度时可直接得到单质汞。但是Ce改性OMS-2催化剂后,该部分过程可被强烈抑制,相比OMS-2催化剂,可显著提高汞的表观氧化效率。
王立刚, 彭苏萍, 陈昌和. 燃煤飞灰对锅炉烟道气中Hg0的吸附特性[J]. 环境科学, 2003,24,(6): 59-62. WANG Li-gang, PENG Su-ping, CHEN Chang-he. The experimental study to Hg0 adsorption of fly ash in flue gas[J]. Environ Sci, 2003, 24(6): 59-62.
任建莉, 周劲松, 骆仲泱, 徐璋, 张雪梅. 钙基类吸附剂脱除烟气中气态汞的试验研究[J]. 燃料化学学报, 2006,34,(5): 557-561. REN Jian-li, ZHOU Jin-song, LUO Zhong-yang, XU Zhang, ZHANG Xue-mei. Ca-based sorbents for mercury vapor removal from flue gas[J]. J Fuel Chem Technol, 2006, 34(5): 557-561.
ZHAO Y X, MICHAEL D M, EDWIN S O, JOHN H P, GRANT E D. Effects of sulfur dioxide and nitric oxide on mercury oxidation and reduction under homogeneous conditions[J]. J Air Waste Manage Assoc, 2012, 56(5): 628-635.
WANG F, WANG H M, ZHANG F, ZHU J W, TIAN G, LIU Y, MAO J X. SO2/Hg removal from flue gas by dry FGD[J]. Int J Min Sci Technol, 2012, 22(1): 107-110.
WANG Y Y, SHEN B X, HE C, YUE S J, WANG F M. Simultaneous removal of NO and Hg0 from flue gas over Mn-Ce/Ti-PILCs[J]. Environ Sci Technol, 2015, 49(15): 9355-9363.
YANG W, LIU Y X, WANG Q, PAN J F. Removal of elemental mercury from flue gas using wheat straw chars modified by Mn-Ce mixed oxides with ultrasonic-assisted impregnation[J]. Chem Eng J, 2017, 326(15): 169-181.
XU H M, QU Z, ZONG X, QUAN F Q, MEI J, YAN N Q. Catalytic oxidation and adsorption of Hg0 over low-temperature NH3-SCR LaMnO3 perovskite oxide from flue gas[J]. Appl Catal B: Environ, 2016, 186(5): 30-40.
ATRIBAK I, BUENO L A, GARCIA G A, NAVARO P, FRIAS D, MONTES M. Catalytic activity for soot combustion of birnessite and cryptomelane[J]. Appl Catal B: Environ, 2010, 93(3/4): 267-273.
DING Y S, SHEN X F, SITHAMBARAM S, GOMEZ S, KUMAR R, CRISOSTOMO V M B, SUIB S L, AINDOW M. Synthesis and catalytic activity of cryptomelane-type manganese dioxide nanomaterials produced by a novel solvent-free method[J]. Chem Mater, 2005, 17(21): 5382-5389.
HUANG W M, SHI J L. Water-promoted low-concentration NO removal at room temperature by Mg-doped manganese oxides OMS-2[J]. Appl Catal A: Gen, 2015, 507(25): 65-74.
GANDHE A R, JEANETTE S, REBELLO J, FIGUEIREDO L, FERNANDES J B. Manganese oxide OMS-2 as an effective catalyst for total oxidation of ethyl acetate[J]. Appl Catal B: Environ, 2007, 72(1/2): 129-135.
ADJIMI S, GARCIA V J, DIAZ J A, RETAILLEAU L, GIL S, PERA T M, GUO Y, GIROIR F A. Highly efficient and stable Ru/K-OMS-2 catalyst for NO oxidation[J]. Appl Catal B: Environ, 2017, 219(15): 459-466.
HERNANDEZ W Y, CENTENO M A, SVETLANA I, PIERRE E, GAIGNEAUXE M, ODRIOZOLA J A. Cu-modified cryptomelane oxide as active catalyst for CO oxidation reactions[J]. Appl Catal B: Environ, 2012, 123(23): 27-35.
LI J F, YAN N Q, QU Z, QIAO S H, YANG S J, GUO Y F, LIU P, JIA J P. Catalytic oxidation of elemental mercury over the modified catalyst Mn/alpha-Al2O3 at Lower Temperatures[J]. Environ Sci Technol, 2010, 44(1): 426-431.
LIU X, JIANG S J, LI H L, YANG J P, YANG Z Q, ZHAO J X, PENG H Y, KAIMIN S. Elemental mercury oxidation over manganese oxide octahedral molecular sieve catalyst at low flue gas temperature[J]. Chem Eng J, 2019, 356(15): 142-150.
WU S J, KATAYAMA R, UDDIN M A, SASAOKA E, XIE Z M. Study on reactivity of HgO over activated carbon with HCl and SO2 in the presence of moisture by temperature-programmed decomposition desorption mass spectrometry[J]. Energy Fuels, 2015, 29(10): 6598-6604.
李扬, 刘冰, 杨赫, 杨大伟, 胡浩权. MnOx-TiO2吸附剂对燃煤烟气中汞的脱除[J]. 燃料化学学报, 2020,48,(5): 513-524. LI Yang, LIU Bing, YANG He, YANG Da-wei, HU Hao-quan. Removal of elemental mercury(Hg0) from simulated flue gas over MnOx-TiO2 sorbents[J]. J Fuel Chem Technol, 2020, 48(5): 513-524.
WU S J, UDDIN M A, NAGANO S, MASAKI O, SASAOKA E. Fundamental study on decomposition characteristics of mercury compounds over solid powder by temperature-programmed decomposition desorption mass spectrometry[J]. Energy Fuels, 2011, 25(2): 144-153.
HE S, ZHOU J S, ZHU Y Q, LUO Z Y, NI M J, CEN K. Mercury oxidation over a vanadia-based selective catalytic reduction catalyst[J]. Energy Fuels, 2009, 23(1): 253-259.
赵莉, 何青松, 刘宇, 吴洋文, 陆强, 刘松涛, 董长青. 湿法脱硫浆液中Hg2+的还原机制研究[J]. 燃料化学学报, 2017,45,(6): 755-760. ZHAO Li, HE Qing-song, LIU Yu, WU Yang-wen, LU Qiang, LIU Song-tao, DONG Chang-qing. Mechanism of Hg2+ reduction in wet flue gas desulfurization liquors[J]. J Fuel Chem Technol, 2017, 45(6): 755-760.
LI H L, WU C Y, LI Y, ZHANG J. CeO2-TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas[J]. Environ Sci Technol, 2011, 45(17): 7394-400.
WANG T, LIU J, YANG Y H, SUI Z F, ZHANG Y S, WANG J W, PAN W P. Catalytic conversion of mercury over Ce doped Mn/SAPO-34 catalyst: Sulphur tolerance and SO2/SO3 conversion[J]. J Hazard Mater, 2020, 381(5): 120986.
王艳坤. HSC Chemistry软件在高校化学科研中的应用[J]. 河南教育学院学报:自然科学版, 2013,22,(2): 32-34. WANG Yan-kun. Application of HSC chemistry software in university chemical scientific research[J]. J Henan Ins Edu Nat Sci Ed, 2013, 22(2): 32-34.
GRANITE E J, PENNLINE H W, HARGIS R A. Novel sorbents for mercury removal from flue gas[J]. Ind Eng Chem Res, 2000, 39(4): 1020-1029.
WU Z B, JIANG B Q, YUE L, ZHAO W R, GUAN B H. Experimental study on a low-temperature SCR catalyst based on MnOx/TiO2 prepared by sol-gel method[J]. J Hazard Mater, 2007, 145(3): 488-494.
LI H L, ZHU L, WANG J, LI L Q, SHIH K. Development of nano-sulfide sorbent for efficient removal of elemental mercury from coal combustion fuel gas[J]. Environ Sci Technol, 2016, 50(17): 9551-9557.
HOU J T, LI Y Z, MAO M Y, ZHAO X J, YUE Y Z. The effect of Ce ion substituted OMS-2 nanostructure in catalytic activity for benzene oxidation[J]. Nanoscale, 2014, 6(24): 15048-15058.
LIU C, CHEN L, LI J, MA L, ARANDIYAN H, DU Y, XU J, HAO J. Enhancement of activity and sulfur resistance of CeO2 supported on TiO2-SiO2 for the selective catalytic reduction of NO by NH3[J]. Environ Sci Technol, 2012, 46(11): 6182-6189.
WANG X Y, LAN Z X, ZHANG K, CHEN J J, JIANG L L, WANG R H. Structure activity relationships of AMn2O4 (A=Cu and Co) spinels in selective catalytic reduction of NOx: Experimental and theoretical study[J]. J Chem Phys C, 2017, 121(6): 3339-3349.
KIVELSON, DANIEL. The determination of the potential constants of SO2 from centrifugal distortion effects[J]. J Chem Phys, 1954, 22(5): 904-908.
王鹏鹰.铝基SCR催化剂催化氧化烟气中汞的实验与机理研究[D].武汉: 华中科技大学, 2014.WANG Peng-ying. A dissertation submitted in partial fulfllment of the requirements for the degree of doctor of philosophy in engineering[D]. Wuhan: Huazhong University of Science and Technology, 2014.
张旭楠. CeO2改性SCR催化剂同时脱除烟气中Hg0和NO的实验研究[D].长沙: 湖南大学, 2015.ZHANG Xu-nan. The experimental study on the simultaneous removal of elemental mercury (Hg0) and NO from flue gas by CeO2 modified SCR catalysts[D]. Changsha: Hunan University, 2015.
ZHUANG Y, JASON L, RICHARD L, MIKE H, JOHN P. Impacts of acid gases on mercury oxidation across SCR catalyst[J]. Fuel Process Technol, 2007, 88(10): 929-934.
LAUDAL D L, THOMPSON J S, PAVLISH J H, BRICKET L, CHU P, SRIVASTAVA R K, LEE C W, KILGROE J. Mercury speciation at power plants using SCR and SNCR control technologies[J]. Electron Manager, 2012, 53(1): 16-22.
SUI Z F, ZHANG Y S, LI W H, WILLIAM O, CAO Y, PAN W P. Partitioning effect of mercury content and speciation in gypsum slurry as a function of time[J]. J Therm Anal Calorim, 2015, 119(3): 1611-1618.
ZHANG Y S, ZHAO L L, GUO R T, WANG J W, CAO Y, WILLIAM O, PAN W P. Influences of NO on mercury adsorption characteristics for HBr modified fly ash[J]. Int J Coal Geol, 2017, 170(1): 77-83.

图 1 SO2对催化剂上Hg0氧化性能的影响
Figure 1 Influence of SO2 on the catalyst performance in the oxidation of Hg0

图 2 催化剂表征谱图
Figure 2 Characterization of the catalysts
(a)XRD,(b)Mn 2p XPS能谱图,(c)Ce 3d XPS能谱图,(d)O 1s XPS能谱图
(a): XRD patterns; (b): Mn 2p XPS spectra; (c): Ce 3d XPS spectra; (d): O 1s XPS spectra

图 5 OMS-2催化剂表面SO2对HgO的还原
Figure 5 Reduction of HgO by SO2 on the surface of OMS-2 catalyst

图 6 Ce-OMS-2催化剂表面SO2对HgO的还原
Figure 6 Reduction of HgO by SO2 on the surface of Ce-OMS-2 catalyst

图 8 SO2气氛下各平衡相含量随温度的变化关系
Figure 8 Equilibrium contents of various species in the presence of SO2 at different temperatures Initial contents of HgO and SO2 are set as 1 and those of other species as 0
表 1 实验条件
Table 1. Reaction test conditions
| Test | Catalyst | Carrier gas | Temp. t/℃ | Flow /(L·min-1) |
| 1 | OMS-2 | N2,N2+0.05%SO2,N2+0.1%SO2,N2+0.1%SO2+4%O2 | 150 | 1 |
| 2 | Ce-OMS-2 | N2,N2+0.05%SO2,N2+0.1%SO2,N2+0.1%SO2+4%O2 | 150 | 1 |
| 3 | Ce-OMS-2#1 | N2 | 100-700 | 0.5 |
| Ce-OMS-2#2 | ||||
| 4 | OMS-2* | N2+0.05%SO2 | 150 | 1 |
| 5 | Ce-OMS-2* | N2+0.05%SO2 | 150 | 1 |
| 6 | 10 mg HgO | N2+0.05%SO2 | 150 | 1 |
| note:“#1”, the catalyst was pretreated under N2+4%O2+Hg0 for 1 h; “#2”, the catalyst was pretreated under N2+4%O2+0.05%SO2+Hg0 for 1 h; “*”, the catalyst pretreated under N2+4%O2+500 μg/m3 Hg0 for 7 d | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 催化剂的比表面积和表面原子含量
Table 2. Surface area and surface atomic concentration of the catalysts
| Catalyst | Surface area A/(m2·g-1) | Surface atom concentration | ||||||
| by ICP | by XPS | |||||||
| K/Mn | Ce/Mn | Mn3+/Mn4+ | Ce3+/Ce4+ | Oα | Oβ | |||
| OMS-2 | 173 | 0.10 | - | 1.07 | - | 79.6% | 20.4% | |
| Ce-OMS-2 | 390 | 0.038 | 0.35 | 1.10 | 0.24 | 68.2% | 31.8% | |
| note: Oα: lattice oxygen; Oβ: surface adsorbed oxygen | ||||||||
下载: 导出CSV
表 3 HgO在SO2气氛下的初始工况
Table 3. Initial conditions of HgO under SO2 atmosphere
| Species | HgO | SO2 | O2 | SO3 | Hg | HgSO4 |
| Amount of substance /kmol | 1 | 1 | 0 | 0 | 0 | 0 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们