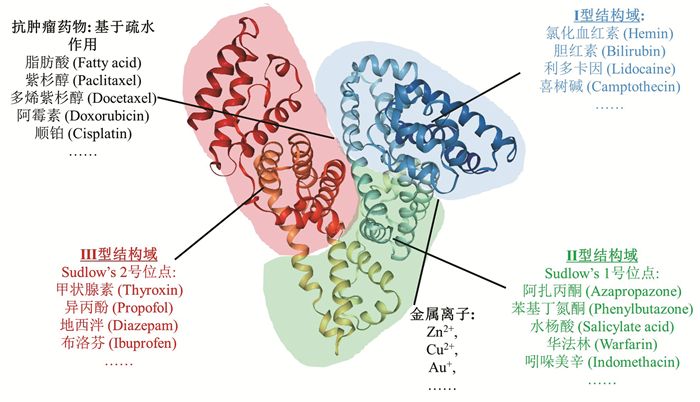
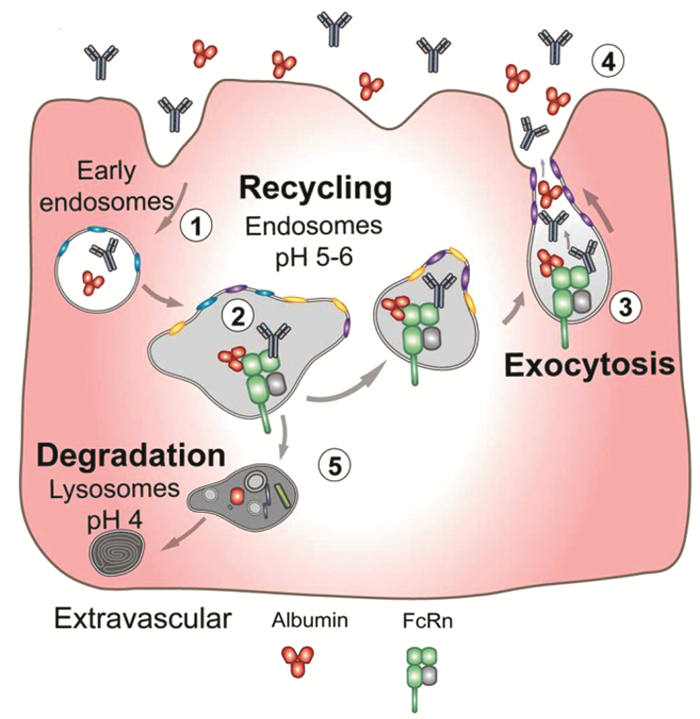
纳米载体在生物医药领域有着广泛的应用,然而现有的纳米载体普遍存在生物相容性差、潜在的毒副作用以及难以被降解、代谢等问题[1 。因此,如何选择与构建高生物相容度的生物纳米载体,已成为现今肿瘤诊疗领域一项极具挑战的课题。血清白蛋白(Serum albumin)是血液里最主要的蛋白质,在维持胶体渗透压和调解血浆pH方面发挥着重要作用[2 。血清白蛋白共有585个氨基酸,晶体研究表明,这些氨基酸组成了三个立体结构类似的结构域,分别为Ⅰ型结构域、Ⅱ型结构域和Ⅲ型结构域,而每个结构域中又分别包含两个次级结构域,即结构域A和次结构域B[3 。部分生理物质[4 (脂肪酸、胆固醇、胆红素)、金属离子(如锌离子、铜离子)以及药物分子(如华法林(Warfarin)、布洛芬(Ibuprofen)、紫杉醇(Paclitaxel,PTX)等)均能插入血清白蛋白不同结构域的腔体(如Sudlow’s 1号位点和Sudlow’s 2号位点)或通过疏水作用以及静电作用与蛋白非共价结合(图 1 [5 。该特点与多种细胞中表达的特殊蛋白受体——新生儿Fc受体(Neonatal Fc Receptor,FcRn)的调节密不可分。FcRn[6 ,又称Brambell受体,是主要组织相容性复合体(Major histocompatibility complex,MHC)1类膜蛋白,能够特异性结合免疫球蛋白(Immunoglobulin G,IgG)和血清白蛋白。FcRn主要分布在胞内体(Endosomes)中,在胞内体酸化(endosomal acidification, < pH 6.5)的条件下会触发FcRn与细胞内的血清白蛋白发生特异性结合,并在pH中性条件下将血清白蛋白重新释放至细胞外;其他未能与FcRn结合的蛋白将进入溶酶体中降解(图 2
图 1
图 2
血清白蛋白的结构学特点、超长半衰期以及其良好的生物相容性使其成为构建生物纳米载体的优异材料。一方面,研究者将血清白蛋白发展为药物载体,从而达到促进药物体内分布,延长药物在体内半衰期以及改善药效动力学等效果。另一方面,研究者通过对血清白蛋白的共价或非共价修饰,赋予其肿瘤成像标记以及协同治疗等多重功能。本文就近年来多功能化血清白蛋白作为载体在肿瘤诊疗领域的进展进行了总结,并探讨了血清白蛋白作为肿瘤诊疗载体的未来发展方向。
2.
血清白蛋白载体的肿瘤诊疗应用
2.1
抗肿瘤药物与血清白蛋白构建复合体
绝大多数抗肿瘤药物存在水溶性差,高毒性以及体内快速分解代谢等缺点[7 9 ,例如PTX[10 作为一种治疗多种癌症的化疗药物,只能依靠聚氧乙烯蓖麻油来溶解,然而约20%~40%的患者会因注射聚氧乙烯蓖麻油导致严重的过敏反应。白蛋白结合型紫杉醇(Abraxane)[11 ,是第一款被美国FDA批准用于临床治疗肿瘤的白蛋白基抗肿瘤药物,PTX通过与白蛋白的结合,能有效解决其水溶性差和致敏问题。此外,研究发现,由于肿瘤细胞表面过表达血清白蛋白结合蛋白,如清蛋白激活蛋白/糖蛋白60(Albondin/Glycoprotein60,gp60)[12 、酸性富含半胱氨酸分泌性蛋白(Secrete protein acidic and rich in cysteine,SPARC)[13 、糖蛋白18/糖蛋白30(Glycoprotein18/Glycoprotein30,gp18/gp30)[14 等,故血清白蛋白能够长期聚集于肿瘤间隙和进入肿瘤内部[15 。因此,通过共价或非共价的形式,把抗肿瘤药物与血清白蛋白结合,是一种提高药物整体效果的新型策略。
部分天然物质因其疏水性结构导致难以在血液中以游离形式传递,但可通过与血清白蛋白的结合达到体内分布的目的。因此,通过对抗肿瘤药物进行结构修饰,在不破坏药物功能的位点情况下添加天然产物衍生物片段,使药物与血清白蛋白载体发生稳定的结合,可以改善药物的水溶性,同时促进其体内分布,提高药效动力。
胆固醇[16 是能和血清白蛋白稳定结合的天然产物之一,Bienk等[17 通过在小干扰RNA(small interfering RNA,siRNA)片段上修饰胆固醇衍生物,利用胆固醇衍生物与血清白蛋白之间的非共价结合,使血清白蛋白成为siRNA片段的生物载体。实验结果证明,siRNA能够有效地得到血清白蛋白的体内降解保护,相比于siRNA片段约12min的半衰期,修饰一个和两个胆固醇衍生物的siRNA片段的半衰期分别延长至45和71 min,修饰两个胆固醇衍生物的siRNA片段对肝脏肿瘤细胞的基因表达沉默率为28%,而siRNA片段单体沉默率仅为4%。
脂肪酸是生物体脂质的基本成分,能够与血清白蛋白进行结合。Callmann等[18 合成了1, 18-十八烷二酸-紫杉醇(1, 18-Octadecanedioic acid-paclitaxel,ODDA-PTX),并将ODDA-PTX与人血清白蛋白(Human serum albumin,HSA)结合。实验结果表明,ODDA-PTX与HSA之间的稳定结合主要是通过羧酸与HSA的疏水位点发生非共价作用,单个HSA蛋白分子最多能以1:5的结合比例结合ODDA-PTX。ODDA-PTX的药效分析表明,其半衰期约为Abraxane的一倍。此外,相比于Abraxane在肿瘤部位的浓度逐渐降低,ODDA-PTX在肿瘤部位的浓度逐渐升高,意味着ODDA-PTX在肿瘤部位能够持续释放。
铂基药物[19 20 如顺铂(Cisplatin)、卡铂(Carbo-platin)和奥沙利铂(Oxaliplatin)等,是一类被FDA批准用于临床的抗肿瘤药物,然而铂基药物的不稳定性和缺乏肿瘤细胞靶向性影响了其肿瘤治疗效果。Ma等[21 设计了一类顺铂药物,并对其结构进行了双重修饰:一是在铂基药物上修饰了糖基分子,使顺铂药物能够靶向肿瘤细胞上的葡萄糖转运蛋白(Glucose transporters,GLUTs)以及有机阳离子转运蛋白(Organic cation transporters,OCTs),提高顺铂药物的肿瘤靶向性;二是在顺铂药物上修饰了脂肪酸衍生物,借助脂肪酸衍生物和血清白蛋白主体之间的结合,提高顺铂药物的稳定性。
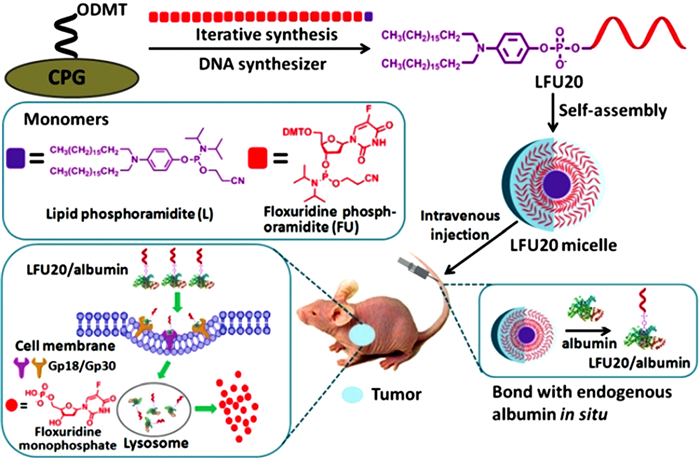
Jin等[22 将1个脂链与20个氟二氧嘧啶通过DNA合成仪合成得到氟二氧嘧啶衍生物(LFU20),LFU20的双亲性结构促使其以胶束形态存在于水中。当LFU20胶束与牛血清白蛋白(Bovine serum albumin,BSA)混合后,因脂链与BSA疏水位点的结合破坏了胶束结构,进而形成LFU20/albumin复合物。实验结果表明,LFU20/albumin能够基于EPR效应(高通透性和滞留效应(Enhanced permeability and retention effect))在肿瘤部位聚集,并在gp18/gp30介导下,进入肿瘤细胞内部,随着溶酶体对血清白蛋白的降解,LFU20逐渐释放从而实现抑制细胞增殖的效果(图 3
图 3
除了天然物质的衍生物能够与血清白蛋白发生稳定结合之外,一些小分子或药物也可以与血清白蛋白的疏水位点发生非共价结合。Wen等[23 将合成的放射性4-(对碘苯基)丁酸([131 I]IBA)与血清白蛋白结合,[131 I]IBA与血清白蛋白形成的复合物具有良好的稳定性,单光子发射计算机断层扫描(SPECT)结果显示,[131 I]IBA在血池、淋巴结以及肿瘤部位均能清晰成像,证明血清白蛋白对[131 I]IBA的体内分布和循环具有促进作用。
喹诺酮(Quinolone)衍生物的还原态在光照下能够释放一氧化碳,进而抑制肿瘤细胞的生长[24 。Popova等[25 将羟基苯并三氮唑喹诺酮(3-Hydroxybenzo[g]quinolone)与BSA通过疏水位点进行非共价结合。实验结果证明,在同等光照条件下,相比于喹诺酮单体,BSA结合的喹诺酮衍生物对肿瘤细胞的毒性更高,并具有一定的肿瘤靶向性。Qi等[26 将抗肿瘤药物5-氟二氧嘧啶(5-Fluorouracil,5-FU)、金属药物2-苯甲酰嘧啶胺基硫脲铜(2-benzoylpyridine thiosemicarbazide copper Ⅱ,BpT)、基因药物AS1411同时与血清白蛋白结合,其中5-FU和BpT通过非共价结合至血清白蛋白主体的疏水位点,AS1411则通过半胱氨酸(cysteine-34,34-Cys)与血清白蛋白发生共价偶联。细胞毒性实验表明,该复合药物体系的细胞毒性约降低7倍,并且血清白蛋白能够提高肿瘤靶向性。
白蛋白结合域(Albumin binding domain,ABD)[27 28 是一种白蛋白结合多肽序列,具有三个螺旋结构,对于血清白蛋白具有高亲和力。将药物分子与ABD通过基因手段融合,可实现与血清白蛋白的稳定结合,进而改善药物治疗效果。Wang等[29 将链球杆菌G蛋白(Streptococcal protein G)衍生的ABD与人铁蛋白(Human ferritin,HFn)进行融合形成ABD-HFn纳米颗粒,并通过HFn负载抗肿瘤药物阿霉素(Doxorubicin,DOX)。实验表明,基于ABD与血清白蛋白的亲和性,ABD-HFn纳米颗粒所负载的DOX半衰期延长至17.2h,分别是参照物DOX和利用未融合ABD的HFn负载DOX的19倍和12倍。
Yousefpour等[30 将基因编码G蛋白衍生物的ABD与嵌合蛋白(Chimeric polypeptide,CP)融合形成ABD-CP,进一步通过ABD-CP的C端与DOX共价偶联形成DOX-ABD-CP。实验表明,注射至体内后,DOX-ABD-CP能够与血清白蛋白自组装形成血清白蛋白胶束,从而延长DOX在血液循环中的时间,提高在肿瘤部位的累积。他们[31 还利用基因合成技术将双性离子型(Zwitterionic)多肽KEKE、弹性样多肽(Elastin-like polypeptide,ELPs)、分选酶A剪切位点(Sortase A cleavage site,srt)、ABD以及半胱氨酸肽链(CGG)依次连接,再将DOX与CGG末端共价偶联,最终形成KEKE-ELPs-srt-ABD-DOX,将其在大肠杆菌中进行表达,并通过分选酶A对srt位点的剪切,从而纯化出ABD-DOX。ABD-DOX与DOX之间的活体实验结果表明,DOX在小鼠体内约15min后被分解代谢,而ABD-DOX的半衰期约为29.4h。注射后2h对药物在肿瘤部位的浓度进行检测,ABD-DOX的浓度约为DOX的4倍,而且ABD-DOX在肿瘤中能稳定维持72h,其相对累积浓度约为DOX单体的120倍。由于ABD-DOX具有更高的药效动力学和体内分布,其对肝癌和胰腺癌有更好的治疗效果。
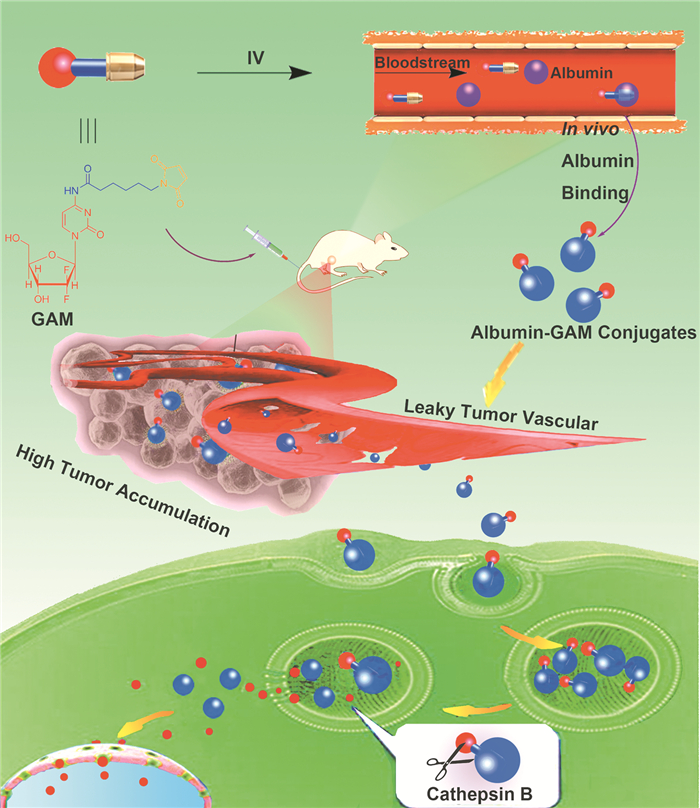
由于血清白蛋白的34-Cys上的巯基未形成双硫键,因此具有很强的反应活性[32 。药物分子与血清白蛋白主体的结合,可通过与血清白蛋白的活性反应位点共价偶联来实现。Mullapudi等[33 通过对膀胱癌细胞UMUC3的分析,筛选出具有靶向UMUC3细胞过表达CD47的多肽txCD47,将其与血清白蛋白通过巯基反应偶联,再利用血清白蛋白结合抗肿瘤药物多西他赛(Docetaxel,DTX),构建形成txCD47-ABD-DTX复合体系。实验表明,相比于DTX单体,txCD47-ABD-DTX的肿瘤细胞毒性IC50 值高出一个数量级,同时该体系能借助血清白蛋白进入膀胱癌肿瘤细胞,从而避免了随尿液快速排泄,其在膀胱中的滞留时间超过72h。He等[34 将含有马来酰亚胺基团的吉西他滨(Gemcitabine,GEM)衍生物与血清白蛋白的34-Cys在血液中进行原位反应,获得血清白蛋白与GEM的聚合物Albumin-GAM,这类新型药物具有更好的肿瘤靶向性,同时该药物存在有对组织蛋白酶B(Cathepsin B)敏感的连接键,当药物进入到肿瘤内部后,可通过酶剪切促使药物释放(图 4
图 4
虽然药物分子与血清白蛋白共价或非共价结合能够延长半衰期和获得更好的体内分布,但是药效动力学并未一定得到改善,例如血清白蛋白的内部结构可能会影响药物与受体之间的结合。胰高血糖素类多肽[35 (Glucagon-like peptide 1,GLP-1)能够提高胰岛素响应,然而其约3min的半衰期限制了其临床应用,Bukrinski等[36 将GLP-1与重组HSA(recombinant HSA,rHSA)进行融合,实验表明GLP-1与rHSA结合后其半衰期时间明显延长,但由于rHSA的空间位阻,致使GLP-1的药效下降了3~4个数量级。此外,血清白蛋白单体的结合位点以及结合能力均是有限的,血清白蛋白结合的药物效果仍受到剂量影响。因此,构建血清白蛋白纳米颗粒能够提高血清白蛋白的结合能力,进而改善药效,并基于纳米颗粒的结合方式和立体结构,获得降低药物毒性和可控释放药物等功能。
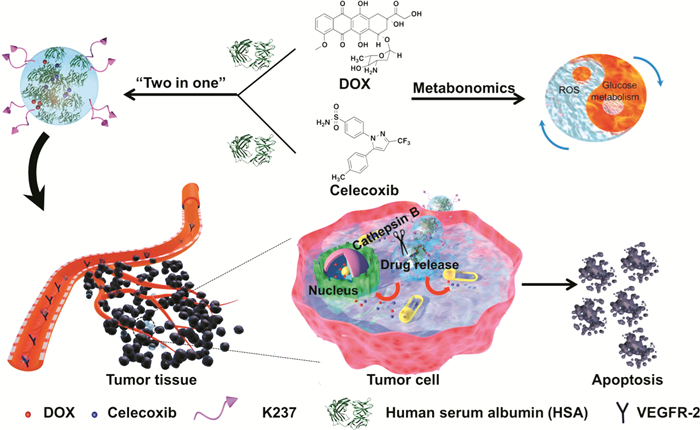
Shi等[37 将HSA作为生物主体,将DOX与2型环氧化酶(Cyclooxygenase 2,COX-2)抑制剂塞来昔布(Celecoxib)分别与HSA非共价结合,在塞来昔布的诱导下,两种HSA能够自组装为纳米颗粒,为进一步提高HSA纳米颗粒的肿瘤靶向性,在HSA上通过点击化学反应修饰具有靶向细胞血管内皮生长因子(Vascular endothelial growth factor receptor 2,VEGFR-2)的肽链片段K237。这一纳米颗粒基于塞来昔布与DOX之间的协同作用,能够显著提高对肿瘤细胞的治疗效果(图 5
图 5
Hu等[38 发现铜离子(Cu2+ )与5-硝基-8-羟基喹啉(5-nitro-8-hydroxyquinoline,NQ)形成的金属有机物Cu(NQ)2 具有一定的抗肿瘤效果,但Cu(NQ)2 的水溶性极差,因此借助与BSA之间的结合构建形成的Cu(NQ)2 -BSA纳米颗粒,不仅能提高水溶性和肿瘤靶向能力,而且相比于NQ或Cu(NQ)2 ,具有更强的肿瘤细胞毒性。Lin等[39 将抗肿瘤药物PTX或芬维A胺(Fenretinide)与BSA结合,BSA会在疏水性药物的作用下自组装为纳米颗粒,进一步将细胞渗透多肽序列低分子量精蛋白(Low molecular weight protamine,LMWP)与BSA纳米颗粒共价偶联。实验证明,在血清白蛋白受体蛋白gp60的介导下,BSA纳米颗粒能够突破血脑屏障,对神经瘤细胞起到抑制作用。Ai等[40 将甘露糖化的阳离子BSA构建为纳米颗粒,并利用纳米颗粒对脱氧寡糖核苷核酸(Oligodeoxynucleotides,ODNs)进行负载。实验证明,基于甘露糖与巨噬细胞表面过表达的受体之间特异性的结合,BSA纳米颗粒能够大量进入巨噬细胞,其所负载的ODNs虽然未影响肿瘤细胞的整体活性,但能够提高促炎因子(Proflammatory cytokines)的分泌,从而减弱因肿瘤导致的免疫抑制。
Razi等[41 将还原态BSA与乙二醇化壳聚糖通过疏水作用及双硫键构建纳米颗粒rBG-NPs,并利用纳米颗粒封装PTX。实验表明,纳米颗粒经细胞内吞作用进入细胞后对PTX开始缓慢释放,共同孵育48h后其细胞毒性逐渐高于PTX单体。Gad等[42 通过普朗尼克(Pluronic)F127、DTX以及HSA构建复合纳米晶体F127-DTX@HSA。实验结果表明,F127-DTX@HSA可基于EPR效应聚集于肿瘤部位,同时由于耐药肿瘤细胞表面能够表达更多的SPARC,纳米晶体能够更容易进入耐药肿瘤细胞。Gong等[43 通过碱性试剂诱导血清白蛋白构象变化,使PTX和血清白蛋白聚合为纳米颗粒NPs-PTX。实验表明,NPs-PTX的肿瘤抑制率为60.8%,明显高于PTX单体的31.2%,同时纳米颗粒能够降低PTX对正常细胞的毒性。
血清白蛋白表面存在大量的负电荷,金属离子能够通过静电作用使血清白蛋白聚集,并进一步形成纳米颗粒,这一过程又称为生物矿化[44 。Ai等[45 将HSA与MnCl2 进行混合,随着MnCl2 氧化为MnO2 ,HSA逐渐聚集在MnO2 表面,最终形成HSA包裹的MnO2 纳米颗粒(MnO2 @HSA)。实验表明,将131 I通过颗粒进行负载后,能够有效地被肿瘤细胞摄入,同时肿瘤内的还原环境促使MnO2 分解,从而进一步释放131 I。Tian等[46 以铈离子(Ce3+ )为核心,以生物矿化的形式构建Ce-BSA纳米材料,通过调节两者的反应时间、摩尔比例以及温度,可形成Ce-BSA纳米链、Ce-BSA纳米簇以及Ce-BSA纳米颗粒。研究发现,Ce-BSA纳米材料具有超氧化物歧化酶的相似活性。Yang等[47 以锌离子与2-甲基咪唑构建ZIF-8金属有机骨架,再以生物矿化机制将BSA封装于ZIF-8内部,构建BSA@ZIF-8纳米颗粒。为提高BSA@ZIF-8纳米颗粒的稳定性,保护蛋白避免被酶降解,利用聚乙烯吡咯烷酮(PVP)对纳米颗粒进行包裹,在肿瘤弱酸性条件下,PVP及ZIF-8会快速分解释放出BSA。
通过聚合物与血清白蛋白的复合构建形成的纳米颗粒,能够提高蛋白的稳定性,改善药效动力学[48 。Saha等[49 通过月桂胺与BSA发生酰胺化反应构建脂肪胺共聚阳离子血清白蛋白纳米颗粒(FCBSA),并通过Cys-34位点结合生物素(Biotin)形成bt-FCBSA。实验证明,负载了DOX的bt-FCBSA能够有效靶向肿瘤细胞上的生物素受体,并释放DOX以促使细胞死亡。Jiang等[50 将聚(低聚甲基丙烯酸乙二醇酯)(POEGMEA)与聚己内酯通过断裂链转移(RAFT)聚合形成POEGMEA-PCL,同时将PCL与BSA反应形成BSA-PCL。POEGMEA-PCL与BSA-PCL能够以不同比例混合构建纳米颗粒,并负载姜黄素实现抑制肿瘤细胞的效果。实验表明,BSA-PCL的比例上升将提高纳米颗粒的细胞毒性和肿瘤细胞选择性。
纳米材料的表面比内部具有更高的能量,因此当纳米材料进入生理环境后,其表面会自发地吸引蛋白分子,形成蛋白冠状物[51 (Protein corona),从而降低纳米材料的表面能,并降低生理系统对纳米颗粒的免疫响应。Cao等[52 设计了类冠状物血清白蛋白纳米颗粒,将九聚精氨酸与二油酰磷脂酰乙醇胺(DOPE)通过双硫键偶联形成双亲性多肽衍生物r9-S-S-DOPE;r9-S-S-DOPE可以与疏水的IR-780组装为DRI,并负载siRNA组装为DRI-S,再利用HSA形成蛋白冠状物最终构建形成HSA包裹的纳米复合系统DRI-S@HSA。DRI-S@HSA能够在扩散性乳腺癌细胞中大量内化,同时在激光照射下表现出显著抑制细胞迁移和增殖的作用。
2.2
多元化诊疗试剂与血清白蛋白构建复合体
血清白蛋白主体与抗肿瘤药物的结合,为肿瘤的化学治疗提供了新的思路,然而绝大多数研究主要还是集中于利用EPR效应的被动靶向或者是血清白蛋白与肿瘤受体之间的结合,从而提高药物对肿瘤的抑制效果。研究发现,肿瘤细胞的生长、增殖、扩散并不具有规律性,单一功能的药物治疗仍存在诸多局限,例如药物未到达肿瘤部位的“脱靶”,或者无法对扩散性肿瘤有效杀伤[53 。因此,利用光、声、电信号对肿瘤细胞进行标记监测,结合放射治疗、光热治疗(Photothermal therapy,PTT)和光动力治疗(Photodynamic therapy,PDT)的诊疗一体化手段,能够获得更好的肿瘤治疗效果。
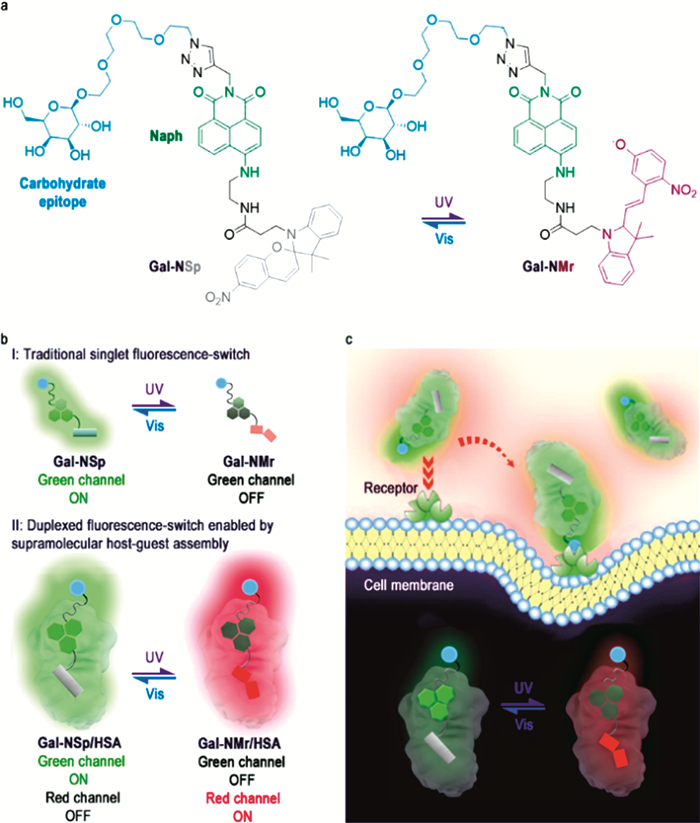
血清白蛋白的疏水位点能够和多种功能成像分子进行结合,并通过信号的放大或变化实现对肿瘤的监测。Fu等[54 报道了一种血清白蛋白与糖基荧光探针的复合物,通过将HSA与半乳糖修饰的萘酰亚胺-螺吡喃(Galactose modified naphthalimide-spiropyran/merocyanine dyad,Gal-NSp/Gal-NMr)/部花青素异构体进行组装,可实现对紫外和可见光比率型响应。光学实验表明,糖基螺吡喃化合物Gal-NSp在紫外照射下会转变为糖基部花青素Gal-NMr,在530nm附近处Gal-NSp化合物的光学信号猝灭,同时Gal-NMr在650nm处的光学信号增强;进一步在可见光照射条件下,Gal-NMr转变为Gal-NSp,在650nm处Gal-NMr的光学信号猝灭,530nm附近Gal-NSp的光学信号增强。然而,Gal-NMr的低量子产率不利于其应用于肿瘤细胞的荧光标记。将HSA作为生物载体,当Gal-NMr进入HSA的疏水位点后,其光学信号显著增强,同时基于HSA的负载,探针能够有效地实现肝癌细胞的荧光标记(图 6 [55 将甘露糖通过二丁基乙炔与荧光素标记的BSA进行偶联,构建具有甘露糖受体靶向性复合体系,利用BSA良好生物相容性及非特异性吸附,可用于检测甘露糖的特定受体如伴刀豆球蛋白A(Concanavalin A,ConA)、大肠杆菌菌毛(pili of E.coli )K12以及肿瘤细胞中的溶酶体。
图 6
除了荧光成像之外,磁共振成像(MRI)[56 也是肿瘤诊断中广泛应用的技术。Zhang等[57 将钆(Gd)离子与富勒烯衍生物组成大分子显影剂,并通过HSA对其负载。实验结果表明,基于HSA和富勒烯之间的协同作用,使显影剂具有更好的稳定性和弛豫率,同时基于HSA的EPR效应,肿瘤部位的显影效果有明显增强。MRI的成像结果与显影剂的性质和剂量具有直接关系,Zhou等[58 基于偶氮染料伊文思蓝(Evans blue,EB)与HSA之间的稳定结合,将T1 加权成像和T2 加权成像显影剂与EB偶联,进一步与HSA结合,构建双模式MRI体系。实验证明,在皮下肿瘤和脑肿瘤部位,双模显影能大幅提高MRI的信噪比(signal-to-noise ratios,SNR)和对比噪声比(contrast-to-noise ratios,CNR),排除假阳性结果。正电子发射计算机断层显像(Positron emission tomography-computed tomography,PET-CT)[59 是一种新型肿瘤诊断技术,能够精确定位病灶,检测肿瘤细胞的转移征兆。Hasler等[60 设计了两类PET-CT显影剂,其中一类显影剂能与血清白蛋白稳定结合。实验结果表明,通过与血清白蛋白结合,显影剂表现出具有更好的稳定性和体内分布效果,可用于肾脏的PET-CT成像,而未与血清白蛋白结合的显影剂,因其稳定性较差会被肝脏分解,从而在肝脏内形成聚集。
肿瘤的有效治疗离不开肿瘤的准确诊断,血清白蛋白主体与肿瘤诊断试剂的结合可以准确地对肿瘤细胞进行标记和示踪,为肿瘤的精准诊疗提供了先决条件。因此,将肿瘤诊断试剂和治疗试剂同时与血清白蛋白结合,赋予血清白蛋白主体诊疗一体化功能,可为现有肿瘤临床治疗提供新的替代手段。肿瘤细胞针对多种抗肿瘤药物表现出多药耐药性(MDR)[61 ,P-糖蛋白(P-glycoprotein,P-gp)会将药物从细胞中泵出,严重影响化学药物的治疗效果。为避免MDR,往往需要将MDR化学逆转剂(Reverse agents,RRA)与化学药物联用,研究发现,血清白蛋白能够有效规避P-gp。Zhang等[62 将BSA、MnO2 和DOX组装形成复合体系BMDN,基于SPARC对BSA的介导,该体系有效克服了肿瘤细胞的耐药性,当进入到肿瘤细胞后,随着溶酶体对BSA的降解,可以释放DOX;同时MnO2 在肿瘤中会还原成Mn2+ ,可作为MRI的显影剂,从而实现肿瘤细胞的治疗和荧光成像一体化。
PTT[63 和PDT[64 是两种新型的肿瘤治疗手段,在特定光源照射下,光敏剂吸收光能转化为热能或产生活性氧(Reactive oxygen species,ROS)进而对肿瘤细胞进行杀伤。Yang等[65 利用偶氮苯将光敏剂Ce6修饰的HSA(HC)和抗癌药物奥沙利铂(Oxaliplatin)修饰的HSA(HO)交联,构建多功能的纳米体系(HCHOA)。由于偶氮苯在还原酶催化下可被还原为苯胺,该反应和低氧环境有很大关系,因此偶氮苯被认为对低氧环境是敏感的。于是此类纳米体系在进入肿瘤细胞后,会因肿瘤细胞的低氧环境而解构为HC和HO,其中HC的Ce6荧光信号会明显增强并在光源照射下产生单线态氧,同时HO将释放出奥沙利铂,从而对肿瘤细胞实现协同抑制作用。
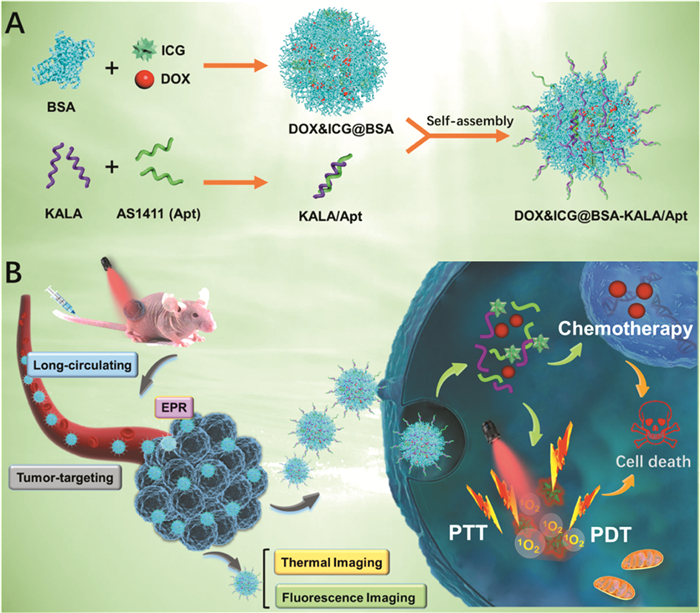
Xu等[66 将抗肿瘤药物DOX和光敏剂ICG通过疏水作用共同负载于BSA上,构建DOX&ICG@BSA纳米颗粒,并在BSA颗粒表面修饰肿瘤适配体(Aptamer)AS1411和细胞穿透肽(Cell-penetrating peptide)KALA以提高纳米颗粒的肿瘤靶向性。实验证明,基于适配体的靶向和纳米颗粒的EPR效应,纳米颗粒能够准确靶向肿瘤并在肿瘤部位长期滞留,随着溶酶体对BSA的降解,在光源照射下ICG的荧光成像、PTT和光动力治疗以及DOX的抗肿瘤功能将同时作用(图 7
图 7
斯托克斯定律认为材料受到高能量的光激发,只能发出低能量的光,比如紫外线激发发出可见光,或者蓝光激发出黄色光,或者可见光激发出红外线。但是研究中发现,有些材料可以实现与上述定律正好相反的发光效果,最具代表性的材料即是上转换发光纳米颗粒(UCNPs)[67 ,其具有自体荧光弱、光稳定性高以及不闪烁等优点。Sabri等[68 利用BSA包裹镧系掺杂UCNPs(NaGdF4:Yb3+ /Er3+ ,Ln-UCNPs),使Ln-UCNPs具备水溶性和生物相容性,同时BSA能够防止水分子的高能量振动以通过偶极-偶极相互作用的方式影响Ln-UCNPs中的光活性离子Yb3+ 和Er3+ ,进而猝灭Ln-UCNPs的荧光;此外,通过在BSA上修饰光敏剂玫瑰红(Rose Bengal,RB),利用Ln-UCNPs的上转换效应,可实现在红外照射下的光学成像和光动治疗功能。
光声成像(Photoacoustic imaging)[69 是指由光激发产生超声信号,当脉冲激光照射到生物组织中时,生物组织产生的超声信号携带了组织的光吸收特征信息,通过探测光声信号能重建出组织中的光吸收分布图像。相比于荧光成像,光声成像结合了纯光学组织成像中高选择性和纯超声组织成像中强穿透性的优点,可得到高分辨率和高对比度的组织图像,从原理上避开了光散射的影响,突破了高分辨率光学成像深度“软极限”(~1mm),可实现50mm深层活体内组织成像。Chen等[70 将Croconine(Croc)染料与HSA自组装构建形成HSA-Croc纳米颗粒,基于Croc上硝基的吸电子性质以及分子内电荷转移机制(ICT),随着纳米颗粒进入肿瘤后pH的降低,Croc的吸收波长将从碱性条件下的680nm红移到酸性条件下的790nm;同时在810nm近红外激光的照射下,随着pH的降低,光热效应逐渐增强,从而实现对肿瘤细胞的光声成像及光热治疗。
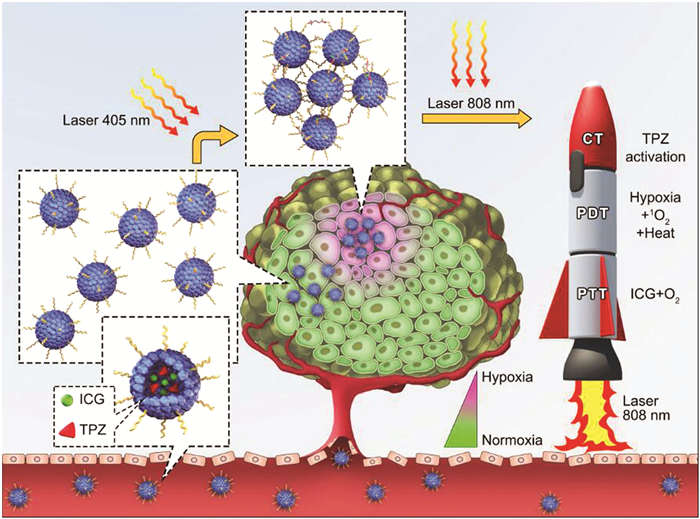
随着血清白蛋白纳米体系构建技术的不断成熟,“All-in-One”策略在血清白蛋白诊疗一体化中得到广泛应用。Han等[71 将氧化钆、金纳米簇、光敏剂ICG与血清白蛋白构建形成纳米复合体系,该复合体系可以基于氧化钆进行MRI和X射线进行成像,同时金纳米簇具有荧光信号以及在光照下释放单线态氧,ICG在光照下可转化热能,从而同时实现集荧光/MRI/X射线/光热/光动五种功能于一体的效果。Hua等[72 将BSA、碳纳米点、铜离子、钆离子以及光敏剂共同构建成复合纳米体系,该体系能够利用荧光、光声以及MRI三种模式进行生物成像,同时具备PDT和PTT作用。实验证明,碳纳米点与光敏剂在光源照射下具有68.4%的光热转化率,远高于单一光热材料,该纳米体系基于EPR效应聚集于肿瘤部位,对肿瘤细胞进行有效抑制。Chen等[73 将光敏剂ICG和抗肿瘤药物替拉扎明(Tirapazamine,TPZ)通过静电作用和疏水效应共同负载于HSA上,形成ICG/TPZ@HSA NMs,将紫光响应交联剂NHS-DA与HSA表面的氨基发生反应,形成复合纳米体系ICG/TPZ@HSA dNMs。在405nm激光照射下,双吖丙啶基团会转变为活性的卡宾基团,从而通过C-H键、C-C键和O-H键与邻近的ICG/TPZ@HSA dNMs发生共价作用,使ICG/TPZ@HSA dNMs聚集成为更大尺寸结构,以提高在肿瘤位点的聚集和延长滞留时间;再通过805nm的激光照射,发挥PTT、PDT和化学治疗的协同作用(图 8
图 8
2.3
肿瘤诊疗试剂与血清白蛋白复合体的活体实验
基于血清白蛋白对于各种肿瘤诊疗试剂的负载效应,肿瘤诊疗试剂的抗肿瘤效果评价不再局限于体外细胞层面。借助血清白蛋白对肿瘤诊疗试剂的降解保护及肿瘤靶向能力,这些蛋白复合体被进一步用于动物实验。脑部肿瘤具有极高的致死率,临床上约98%的小分子药物因无法通过血脑屏障而导致对于脑部肿瘤的治疗效果不佳。缩氧硫脲(Thiosemicarbazones)类分子在其N-3位点的修饰能够有效提高亲脂性以及抗癌活性,Zhang等[74 基于铜基缩氧硫脲分子与HSA之间的疏水作用,利用HSA对铜基缩氧硫脲分子进行保护,并将低分子量精蛋白(LMWP)与HSA共价偶联,构建HSA-C-LMWP复合物。活体荧光成像显示,由于LMWP对于血脑屏障的穿透效力,相比于铜基缩氧硫脲单体,铜基缩氧硫脲与HSA的复合物能更好地富集于神经胶质瘤部位,并促使神经胶质瘤细胞的凋亡率从14.89%提升至38.52%。
原癌基因(K-Ras)的突变与胰腺癌以及结肠癌肿瘤的形成具有直接的关系,同时原癌基因的突变还伴随着巨胞饮(Macropinocytosis)过程的强化。Wang等[75 将抗表皮生长因子(anti-EGFR)与脱辅基蛋白(LDP)以及血清白蛋白进行连接,构建抗肿瘤蛋白药物,其中anti-EGFR可靶向胰腺癌肿瘤细胞,烯二炔荧光团(AE)修饰的LDP具有细胞毒性。活体实验结果表明,HSA能有效延长药物的体内半衰期,药物的荧光信号在注射24h后达到峰值,并能够在肿瘤部位维持360h;而且在用药剂量为0.4mg/kg的条件下,其抑制肿瘤生长效率约81%。
三阴性乳腺癌(Triple-negative breast cancers,TNBCs),因缺少激素受体和人表皮生长因子的表达,因此难以预先诊断,存在很大概率的恶性转移。Liu等[76 通过双硫键构建HSA纳米颗粒以负载PTX,并在纳米颗粒上修饰三阴性乳腺癌甲酰多肽受体的肽链。活体实验证明修饰了靶向肽链的HSA纳米颗粒在注射24h后能够聚集于肿瘤部位,而不是被肝脏代谢,又因HSA对于PTX的保护,负载了PTX的HSA纳米颗粒表现出更强的肿瘤抑制能力(图 9 [77 合成了一种天然产物竹红菌素(Hypocrellin)衍生物AETHB,并通过HSA纳米颗粒的负载提高其水溶性和生物相容性,进而应用于小鼠活体的抗肿瘤研究。结果表明,由于AETHB同时具有PDT和PTT两种治疗功能,在671nm波长的光照下,注射后4h其单线态氧量子产率为0.64,同时瘤体温度从36.5℃升至59.6℃,在PDT和PTT的协同作用下,肿瘤细胞的抑制率约为100%。
图 9
放射治疗是临床治疗肿瘤的一种主要手段,然而现有的放射试剂会在体内快速代谢,而且其会因肿瘤内部缺氧的影响难以达到理想的治疗效果。Tian等[78 将HSA与MnCl2 以生物矿化的策略生成纳米颗粒,并负载标记131 I,构建131 I-HSA-MnO2 纳米体系。活体实验结果表明,这一体系能够有效地被肿瘤细胞摄入,并在肿瘤内部的还原条件下使MnO2 分解,形成131 I-HSA。此外,由于MnO2 与肿瘤内部的H2 O2 可反应生成氧气,相比于131 I单体对于肿瘤生长的部分抑制,131 I-HSA-MnO2 可直接破坏肿瘤细胞,表现出更显著的肿瘤抑制效果。
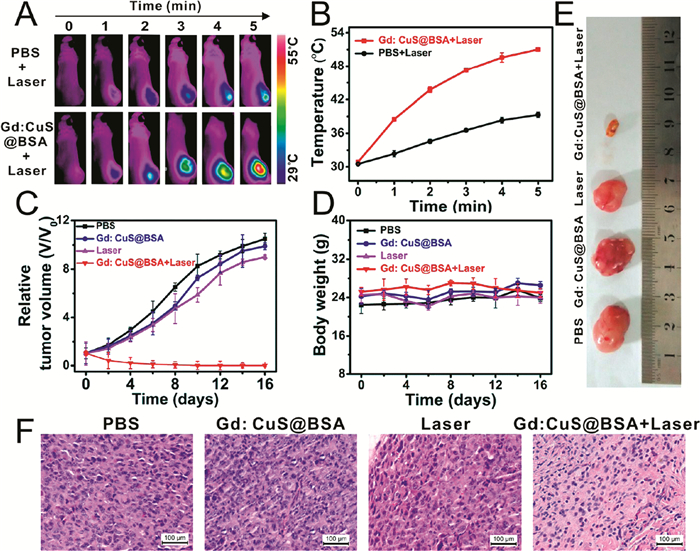
肿瘤微环境(Tumor microenvironment,TME)是由多种细胞构成的复杂组织,其主要特点是缺氧、弱酸性及存在过量的过氧化氢。由于肿瘤细胞的扩散倾向于寻找类似的生理环境,因此TME对于肿瘤的增殖至关重要。Chen等[79 利用HSA负载光敏剂二氢卟吩(Chlorine e6,Ce6)和抗肿瘤铂基药物(c , t , c -[Pt(NH3 )2 -(O2 CCH2 CH2 COOH)(OH)Cl2 ],(cis -Pt(Ⅳ)SA)),并在碱性条件下加入Mn2+ 进行生物矿化以构建HSA-MnO2 -Ce6&Pt(HMCP)纳米颗粒。HMCP纳米颗粒能够基于EPR效应富集于肿瘤组织内部,又因TME中存在过量的H2 O2 ,进入肿瘤内部的HMCP纳米颗粒会因MnO2 与H2 O2 原位反应而分解为尺寸更小的HCP纳米颗粒。进一步对分解后的HCP纳米颗粒的抗肿瘤效果进行评价,活体实验结果显示,在660nm波长的光照下,Ce6可释放自由基实现光动力治疗,同时结合顺铂药物的化学治疗,可实现在低给药剂量(2.7mg·kg-1 Ce6,3.3mg·kg-1 cis -Pt)条件下的肿瘤抑制。Yang等[80 将Gd3+ 和Cu2+ 与BSA混合构建了BSA包裹的Gd:CuS@BSA纳米颗粒,光声和磁共振双模态成像结果显示,Gd:CuS@BSA纳米颗粒的肿瘤富集程度在注射后24h达到峰值,在980nm波长的红外光照下,小鼠肿瘤部位温度从30℃升至51℃,对肿瘤表现出明显的抑制作用(图 10
图 10
Ren等[81 将全氟三丁胺(PFTBA)与负载了光敏剂IR780的HSA构建核壳结构的PFTBA@IR780-HSA纳米颗粒,IR780的荧光信号显示,注射48h后,PFTBA@IR780-HSA纳米颗粒主要富集于小鼠肿瘤部位,在808nm波长红外光照下,PFTBA能够增强光敏剂释放氧自由基的能力,从而有效抑制肿瘤细胞的生长。Wang等[82 基于EB和血清白蛋白之间的稳定结合,将EB首先通过巯基化修饰于金纳米棒(GNRs)表面,然后将GNRs与负载了羟基喜树碱(HCPT)的HSA进行非共价结合,构建了HCPT/HSA/tEB-GNR(HHEG)复合纳米体系。EB的荧光成像和金纳米棒的光声成像结果表明,HHEG纳米体系能够基于EPR效应有效靶向小鼠肿瘤部位,并在注射后12h达到峰值。进一步将注射HHEG后的小鼠置于808nm波长的光照下,一方面,小鼠肿瘤部位温度从29℃迅速升温至55℃,另一方面,相比于基于GNRs的单一PTT,羟基喜树碱和金纳米棒的联合作用对于肿瘤具有更显著的抑制效果,且对于正常器官只有微小损伤。
3.
结语和展望
近年来血清白蛋白主体在肿瘤诊疗领域的应用已越来越广泛,本文主要就基于血清白蛋白构建的复合体系在诊断和治疗方面的应用及发展进行了总结:
首先,多种抗肿瘤药物可以通过疏水作用、静电作用或共价偶联的形式与血清白蛋白结合,通过血清白蛋白主体对药物的保护,避免生理系统对药物的快速代谢,并减少免疫系统对外源性药物分子的不良响应。此外,基于FcRn受体的介导转运,血清白蛋白能够延长药物的体内循环时间,最终获得改善药效动力学的效果。
其次,多种诊断试剂可以利用血清白蛋白主体进行肿瘤组织或细胞的靶向传递。血清白蛋白可基于EPR效应或肿瘤细胞过表达的蛋白受体在肿瘤部位进行富集,提高诊断试剂对肿瘤组织或细胞的内化能力。同时,部分诊断试剂可通过与血清白蛋白内部疏水位点的相互作用,增强试剂的成像信号,提高肿瘤诊断的准确性。
再是,深入研究表明,进一步借助血清白蛋白的负载或包裹功能,可将诊断试剂与治疗试剂进行一体化整合,从而实现对肿瘤的原位诊断和治疗,一方面多种成像模式能提高对肿瘤的定位,避免假阳性结果或“脱靶”现象的发生;另一方面多种治疗试剂的协同作用能够提高对肿瘤的抑制效率,最大程度实现对肿瘤细胞进行杀伤,从而获得比单一模式更精准的多元化诊疗效果。
虽然血清白蛋白已被作为一种生物载体应用于肿瘤临床治疗,但仍存在一些未解决的问题,对于这些问题的改善以进一步拓展和提升血清白蛋白型肿瘤诊疗试剂的功效,是血清白蛋白作为生物载体的重要发展方向:
首先,现有诊疗试剂与血清白蛋白结合的主要目的是利用血清白蛋白对试剂进行体内降解保护或延长其半衰期。然而试剂对血清白蛋白本征生物学效应如其代谢、降解机制等的影响却极少有报道。研究发现,gp60受体对天然血清白蛋白具有结合作用,而gp18/gp30主要结合变性或失活的血清白蛋白。与诊疗试剂结合后的血清白蛋白往往会“变性”,并导致其转运通路信号变化,影响其与受体的亲和性。因此,血清白蛋白载体与诊疗试剂之间相互作用的分子基础仍需进一步精细化研究和评价。
其次,血清白蛋白纳米颗粒的构建方式目前主要依赖于共价交联或金属离子参与的生物矿化,然而这些方法难以精确控制血清白蛋白纳米颗粒的尺寸、形貌和立体结构。因此,发展精确可控的血清白蛋白纳米颗粒制备方案,将为此类纳米材料的临床转化提供重要依据。例如,对血清白蛋白Cy34上存在的唯一反应活性巯基进行有序可控的聚合反应,有望获得结构均一的血清白蛋白纳米颗粒。
再是,修饰的血清白蛋白载体虽然能够自发聚集在肿瘤部位并促进内化,但研究发现仍有部分肿瘤细胞是血清白蛋白无法进入的,因而需根据不同肿瘤细胞的特点,对血清白蛋白主体的细胞渗透能力进行个体化设计。同时,现有的血清白蛋白型诊疗主体对于抑制远端转移的恶性肿瘤细胞仍存在限制,因此发展免疫治疗或基因治疗手段,通过调动全身免疫系统对肿瘤细胞进行杀伤,或通过对肿瘤细胞基因的修复从根源处抑制肿瘤,是血清白蛋白作为诊疗载体的另一个重要研究方向。
综上所述,发展诊疗一体型血清白蛋白主体、实现精准高效的治疗肿瘤疾病,对于肿瘤临床治疗具有重要的意义,也为其他人类重大疾病的诊疗提供了新型平台,必将推进生物医学和生命科学的进一步发展。

 下载:
下载:


 下载:
下载:











