Table 1.
Approaches for isolation of exosomes.
Citation:
Tianqi Wang, Yanan Fu, Shengjie Sun, Chenyi Huang, Yunfei Yi, Junqing Wang, Yang Deng, Meiying Wu. Exosome-based drug delivery systems in cancer therapy[J]. Chinese Chemical Letters,
2023, 34(2): 107508.
doi:
10.1016/j.cclet.2022.05.022

Exosome-based drug delivery systems in cancer therapy
English
Exosome-based drug delivery systems in cancer therapy
Abstract:
Exosome, which is a kind of extracellular vesicles with size around 40-160 nm, plays an important role in cell-to-cell communication in multiple diseases. Especially in tumor microenvironment, exosomes are the important pathway to transit proteins, nucleic acids and small molecules between different kinds of cells. Based on these characteristics, exosomes are served as both therapeutic agents and drug delivery systems in cancer therapy. In this review, the applications of exosomes as drug delivery systems in cancer therapy were summarized and classified according to the cell source of the exosomes, including normal cells, immune cells and tumor cells. Different modifications of exosomes and drug loading methods were presented. Finally, some challenges that hindered the clinical translation of exosomes were also discussed.
-
Key words:
- Exosome
- / Drug delivery
- / Cancer therapy
- / Immunotherapy
- / Macrophages
- / Tumor cells
-
1. Introduction
Exosome, a subset of extracellular vesicles (EVs), is generated from endosome pathway with size from 40-160 nm. Differences in physiological microenvironment lead to heterogeneity of exosomes in source, size, contents, and functions [1, 2]. Normally, exosomes contain proteins, nucleic acids (mRNA and DNA), and some metabolites [3-5]. Membrane proteins expressed on the exosomes result in different effects on their recipient cells and could also be used for the isolation and purification of different kinds of exosomes. The commonly used isolation methods include ultracentrifugation, density gradient, precipitation kits, ExoChip, immunoprecipitation, ExoSearch chip and acoustic nanofilter (Table 1) [6-9]. These methods are single or multiple used for different cell sources depending on the application of the exosomes [10, 11].
Table 1
Because exosome is a main vehicle for the crosstalk of cells in microenvironments, exosome has been explored for treatment of immune-related diseases, different kinds of cancer, cardiovascular and other diseases in decades of years [12-16]. Especially in cancer, 108 exosome-based treatments have moved to clinical trials, indicating that exosome provides new opportunities for cancer treatments [17-19]. For example, Codiak Biosciences presented two clinical trials on exosomes loaded with STING agonist (NCT04592484) and IL-12 (NCT05156229) for immune activation in cancer. MD Anderson Cancer Center also developed Mesenchymal Stromal Cells-derived Exosomes with KRAS-G12D siRNA (NCT03608631) for the treatment of cancer cells with KRASG12D mutations. Some studies have proved that exosome is a good therapeutic agent and delivery system due to its natural advantages, such as accumulation in acidic tumor microenvironment, specific to cancer types, immune effects, and high stability [1, 20-22]. As a drug delivery system in cancer treatments, exosome possesses more attractive features such as nanosize, biocompatibility, tumor targeting ability and easy modification [23, 24]. In addition, exosome could be rapidly transported from the periphery to the lymph nodes (LNs) through lymphatics, which could be served as a drug delivery system for cancer immunotherapy for enhanced immune stimulation [25, 26].
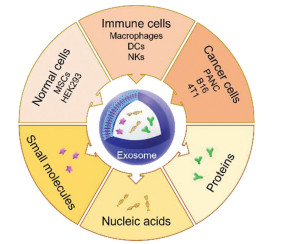
In this review, we will focus on the function of exosomes as drug delivery systems and introduce the application of exosomes derived from different sources for loading small molecular drugs, proteins and nucleic acids in cancer treatment (Fig. 1).
Figure 1
 Figure 1. Scheme of exosomes derived from different sources as drug delivery systems for loading small molecules, nucleic acids and proteins in cancer therapy. MSCs, mesenchymal stem cells; DCs, dendritic cells; NKs, natural killer cells; PANC, pancreatic cell.
Figure 1. Scheme of exosomes derived from different sources as drug delivery systems for loading small molecules, nucleic acids and proteins in cancer therapy. MSCs, mesenchymal stem cells; DCs, dendritic cells; NKs, natural killer cells; PANC, pancreatic cell.2. Normal cell-derived exosomes
Normal cells are more stable and have no inflammatory response [27]. Normal cell-derived exosomes could avoid the clearance in blood circulation and the recognition by reticuloendothelial system (RES). Normal cells have the advantage of stable transfection, indicating that normal cells are good candidates for exosome production and modification. The normally used normal cells include mesenchymal stem cells (MSCs) and human embryonic kidney cells.
2.1 MSC-derived exosomes
MSCs have been used as therapeutic tools in cancer treatment due to their distinctive characteristics including multilineage differentiation, immunosuppression and homing to tumor tissue [28]. Exosome release and paracrine effector are the main pathways for MSCs to perform their biological functions [29]. MSC-derived exosomes (MSC-Exo) were demonstrated to be similar to MSCs on therapeutic effects and could penetrate the tumor tissue, which were conducive to the delivery of therapeutic agents [30]. By single-cell analysis, the distribution of MSC-Exo showed organ-dependent uptake in different cell types and MSC-Exo were more preferred to be uptake by cancer cells in tumor tissue [31]. Moreover, MSCs were easy to be engineered to construct stable expression cell lines, implying that MSC-Exo were the promising tools to design desirable specific cell-targeting delivery systems [32]. Recently, the large-scale generation of functional mRNA encapsulating exosomes via cellular nanoporation demonstrated that MSC-Exo exhibited hyperproductivity and hypoimmunogenicity, indicating that MSC was an ideal cell line to generate exosomes for gene delivery [33].
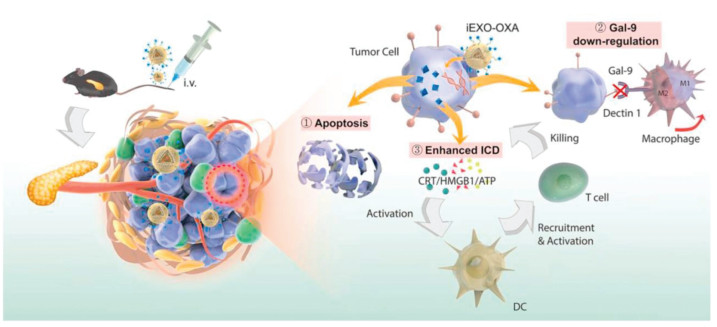
Bone marrow-derived MSCs (BM-MSCs) were used in pancreatic cancer therapy based on the osculating relationship between bone marrow and pancreatic cancer [34, 35]. Exosomes derived from BM-MSCs showed the tumor homing function in (pancreatic ductal adenocarcinoma) PDAC mouse models [36]. Zhou et al. developed a dual delivery system by electroporation loading galectin-9 siRNA and surface modification of oxaliplatin on BM-MSC-derived exosomes [37]. BM-MSC-derived exosomes significantly improved tumor targeting efficacy and elicited antitumor immune responses by M2-type TAM polarization, cytotoxic T lymphocytes recruitment and Tregs downregulation (Fig. 2). In recent years, MSCs derived from other organs such as adipose tissue or stem-like cells such as pediatric diffuse intrinsic pontine glioma cells were also employed as the sources of exosome generation, and theses exosomes were also investigated as vectors for drug delivery in cancer therapy [38, 39].
Figure 2
 Figure 2. Schematic of OXA and galectin-9 siRNA co-loaded delivery system for pancreatic cancer treatment through enhancing immune response and reprogramming tumor microenvironment. Copied with permission [37]. Copyright 2021, Elsevier.
Figure 2. Schematic of OXA and galectin-9 siRNA co-loaded delivery system for pancreatic cancer treatment through enhancing immune response and reprogramming tumor microenvironment. Copied with permission [37]. Copyright 2021, Elsevier.2.2 HEK293 cell-derived exosomes
HEK293 cell is a kind of human embryonic kidney cell line, which is regarded as a safe source for exosome isolation due to the low cancer-related and immunogenically induced mechanisms [40-42]. Based on these characteristics, HEK293 cells were used for producing adenovirus, implying the large-scale production of exosomes for translation [43]. As delivery vehicles, HEK293-derived exosomes had the advantage of easy modification by transfection and other methods for different therapeutic strategies. For example, HEK293 cells were transfected by decoy-binding domain of TNF receptor to display human TNF receptor-1 (hTNFR1) on the surface of exosomes [44, 45]. The engineered exosomes were exploited to co-deliver 5-FU and miR-21 inhibitor oligonucleotide for treatment of Her2 positive cancer and 5-FU resistant colorectal cancer [46]. HEK293 cells transfected by signal regulatory protein α (SIRPα)-plasmid were used to produce SIRPα contained membrane-scaffold-based exosomes for protein delivery [47]. Comparing with protein-scaffold based ferritin nanocages, exosomes exhibited superior antitumor effect due to the unique characteristics, such as providing ideal membrane scaffolds to support the activity and distribution of membrane proteins, which could enhance the phagocytic function of bone marrow derived macrophages (BMDMs) toward cancer cells. Recently, a new technology called EXPLOR, which exploited the natural exosome biogenesis process and reversible protein-protein interactions controlled by optogenetics, was presented for loading soluble proteins into exosomes [48]. This technology was applied to generate srIκB-loaded HEK293T-derived exosomes for sepsis therapy [49]. With the knowledge of protein interactions, EXPLOR will be applied for delivery of proteins in cancer therapy in the future.
3. Immune cell-derived exosomes
Immune cell-derived exosomes preserve part of same functions as their parental cells, such as immune stimulation and inhibition [50]. The potential of immune cell-derived exosomes in cancer therapy depends on the safety and functions of their parental cells in immune responses. Immune cells, including macrophages, dendritic cells (DCs) and natural killer (NK) cells, can be served as the excellent sources for exosome isolation due to their unique features, such as targeting ability and immune activation, which make these exosomes attractive carriers for drug delivery in cancer treatment.
3.1 Macrophage-derived exosomes
Macrophages are heterogeneous cells which could be polarized into different phenotypes in tumor microenvironment during different tumor progression [51]. Macrophages that are activated by lipopolysaccharide (LPS) are tumor suppressive M1-type with tumor inhibiting, tumor vascular normalizing and stromal destruction properties [52]. In tumor tissue, tumor associated-macrophages (TAMs), which exhibit tumor promoting behavior and release some anti-inflammatory cytokines, are similar to M2-type macrophages [53]. Interestingly, M1-type macrophages could secret some molecules to repolarize M2-type macrophages to M1-type [54]. The heterogeneous properties of macrophages lead to different functions of macrophage-derived exosomes. Leaf Huang's group investigated the effect of macrophage phenotypes on the function of exosomes for delivery of lipid calcium phosphate (LCP) nanoparticle-encapsulated Trp2 vaccine (Vac) [55]. The antitumor experiment showed that Vac-M1-Exo exhibited superior antitumor efficiency comparing with Vac-M2-Exo and Vac in vitro and in vivo. Moreover, no significant difference between Vac-M2-Exo and Vac on antitumor efficiency was found. Similar result was verified in another research on investigating the effect of macrophage phenotypes on the function of exosomes for paclitaxel (PTX) loading in cancer therapy [56]. M1-type macrophage-derived exosomes were also able to re-educate TAMs and inhibit tumor growth by reprogramming TAMs into M1-like macrophages and increasing antitumor immunity [57]. These works provided evidences for choosing proper phenotypes when employing macrophages as the exosome donors.
Macrophage-derived exosomes can be isolated from both monocytic cell lines (Raw 264.7 cells, THP-1 cells and J774A.1 cells) and primary cells including monocyte cells. These exosomes could be used as drug delivery systems directly for loading different kinds of therapeutic agents such as small molecule drugs, nucleic acids, and proteins [23, 58-60]. Comparing with free drugs, exosomes could significantly improve the tumor accumulation ability and cellular uptake efficiency, as well as reverse P-gp mediated drug resistance. However, there are still some deficiencies with the native exosomes. In recent years, more and more exosome modification methods have been developed to construct functional exosomes for cancer therapy, which could avoid the insufficient effects of native exosomes and improve delivery efficiency. These modification methods include biological approach, chemical method, and nanotechnology, etc. [61, 62].
To improve the targeting ability to specific cells and avoid the recognition by mononuclear phagocyte system (MPS), macrophage-derived exosomes are modified with targeting ligand or polyethylene glycol (PEG) corona with lipid-based conjugates. 1, 2-Distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol) (DSPE-PEG), which is the normally used lipid for liposome preparation and modification, can be also generalized to exosome modification. Kim et al. incorporated aminoethylanisamide-poly(ethylene glycol)-DSPE (AA-PEG-DSPE) moiety into PTX-loaded exosomes for targeting lung cancer cells [63]. Since macrophages are derived from monocytes, the modification of macrophages-derived exosomes could be initiated from the monocytes. Gong et al. isolated disintegrin and metalloproteinase 15 (A15) expressed exosomes (A15-exo) from continuous protein kinase C activated monocyte-derived macrophages [61]. The A15 was modified as a targeting ligand for specific targeting delivery of doxorubicin (Dox) and cholesterol-modified miRNA 159 (Cho-miR159) to integrin αvβ3-overexpressing tumor cells through RGD-manner. A15-Exo derived from monocyte-derived macrophages was a 2.97-fold higher production yield than that derived from the same number of monocytes, which indicated that macrophages were superb source for exosome production.
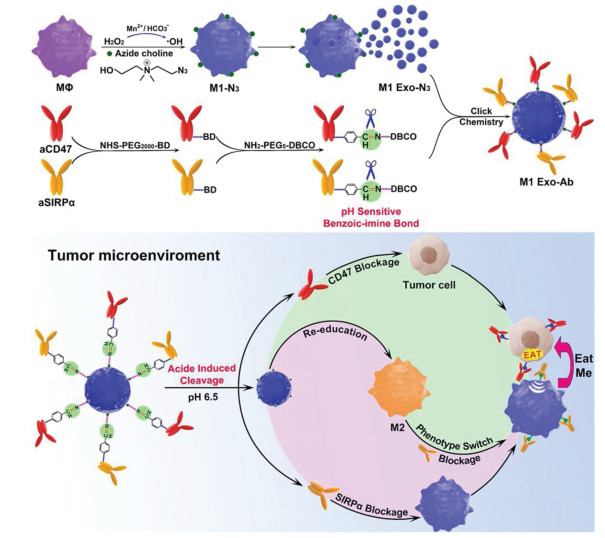
The chemical modification such as click chemistry was also applied to synthesize modified exosomes as nano-bioconjugates for efficient delivery and therapeutic. For example, Nie et al. introduced azide groups to the choline phospholipids on cell membrane to develop aCD47 and aSIRPα modified exosomes with pH-responsive behavior [62]. In tumor microenvironment, the benzoic-imine bonds were cleaved to release two antibodies to improve the phagocytosis of macrophages and polarize M2-type TAM to M1-type for cancer treatment (Fig. 3). In vivo tumor targeting experiment showed that modified M1-Exo exhibited 2.96-fold higher intensity relative to non-modified M1-Exo. Meanwhile, the polarization ability of M1-Exo was not compromised after modification. The click chemistry used in this research could also be generalized to other types of exosomes to improve the targeting ability with minor side effects or modify targeting ligand on the exosome directly. A neuropilin-1 targeted peptide (RGERPPR, RGE) was conjugated to obtain glioma-targeting exosomes for superparamagnetic iron oxide nanoparticles (SPION) and curcumin (Cur) co-delivery [64]. With this engineered exosomes, the diagnostic and therapeutic effects on glioma were significantly improved, while reducing the side effects.
Figure 3
 Figure 3. Schematic of Mn2+-induced M1 macrophage polarization and the synergistic anticancer effect of M1-Exo engineered with aCD47 and aSIRPα. Copied with permission [62]. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 3. Schematic of Mn2+-induced M1 macrophage polarization and the synergistic anticancer effect of M1-Exo engineered with aCD47 and aSIRPα. Copied with permission [62]. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.Nanotechnology was introduced into exosome modification to take full advantage of both exosomes and nanoparticles. Utilizing exosomes as biological membranes to coat the polymeric nanoparticles could improve the biocompatibility and cellular uptake efficiency. Furthermore, the specific cell targeting ligand could be modified on the exosomes to realize specific cell targeting with lower side effect [65]. Rayamajhi et al. developed a hybridized exosome (HE) by extrusion of synthetic liposome and small extracellular vesicles (sEV) and loaded DOX in HE for cancer therapy [66]. HE combined the endogenous and flexible properties of both sEV and liposomes, such as sEV marker proteins, thus endowing it with a higher order of colloidal stability, drug loading, and pH-sensitive sustained drug release.
3.2 Other immune cells
Other immune cells such as DCs, NK cells are also served as the source of exosomes owing to their immune functions in cancer therapy.
DCs, known as the professional antigen-presenting cells (APCs), are involved in the regulation of innate and adaptive immune responses and are associated with cancer progression [67]. The expression of multiple tumor necrosis factor (TNF) superfamily ligands on DCs induced tumor cell apoptosis and activated the antitumor effect of NK cells [68-70]. These surface proteins could be also expressed on DC-derived exosomes (DC-Exo), which indicated that DC-Exo could evoke essential immune functions similar to DCs [71]. Besides TNF, other signature proteins including major histocompatibility complex class Ⅰ (MHC-Ⅰ), class Ⅱ (MHC-Ⅱ) and co-stimulatory molecules could be also expressed on DC-Exo, which preserved most of the functions of their parental cells and was proved to be an effective vaccine in some tumor models [72-76]. Furthermore, DC-Exo was more stable with long self-survival time comparing with DC cells. Thus, immature DC-derived exosomes were regarded as a proper vehicle for therapeutic agents. Non-modified DC-derived exosomes preserved similar antitumor efficiency to chemotherapeutic drugs such as fluorouracil (FU). After loading with FU, FU-DC-Exo was more effective in inhibiting tumor cell proliferation comparing with DC-Exo and FU [77]. To further improve the targeting ability of DC-Exo, lysosome associated membrane glycoprotein 2b (Lamp2b), a well-characterized exosomal membrane protein, was used as a skeleton for confusion of different targeting ligand including the central nervous system-specific rabies viral glycoprotein (RVG) peptide, muscle-specific peptide, and tumor targeting peptide iRGD (CRGDKGPDC) [78-80]. These modified immature DC-Exo could effectively deliver siRNA, Dox and recombinant methioninase to target cancer cells with no overt side effects comparing with non-modified groups. Even though, whether the nucleic acids and proteins in DC-derived exosomes will influence the recipient cells is still unknown and needs to be characterized in the future to enhance the application of these kinds of exosomes.
NK cells, which express some killer proteins (factor related apoptosis ligand (FASL) and perforin), show cytotoxicity in diverse cancer types. Increasing studies have been explored on activated NK cells as a new strategy for antitumor immunity [81]. Meanwhile, NK cell-derived exosomes (NK-Exo) were also demonstrated to be effective antitumor agents in glioblastoma and melanoma [82, 83]. The antitumor effect of NK-Exo was attributed to both the tumor-specific accumulation effect and the killer protein expressed in exosomes [84, 85]. NK-Exo also seemed to be a good candidate for drug or gene delivery in cancer therapy due to these advantages [86-88]. NK-Exo carrying miR-186 could concurrently deliver miR-186 to NK and to cancer cells, retained their killing ability in an immunosuppressive microenvironment and inhibited the growth of neuroblastoma cancer cells [87]. Furthermore, miRNA-loaded dendrimers could also be encapsulated in NK-Exo to form a camouflage biomimetic core-shell structure to avoid the contamination of immunogenic substances [88]. Although NK-Exo presents a specific tumor targeting effect, the mechanism is still not clear and needs to be explored in the future.
4. Tumor cell-derived exosomes
Tumor cells are the most abundant cells in tumor microenvironment and the crosstalk between cancer cells and other types of cells relies on the components of exosomes, including gene, protein, and cytokines, etc. [16, 89, 90]. Tumor cell-derived exosomes are also the widely used vesicles for small molecular drug, gene and protein delivery in cancer therapy [91-94]. The delivery systems based on tumor cell-derived exosomes are listed in Table 2.
Table 2
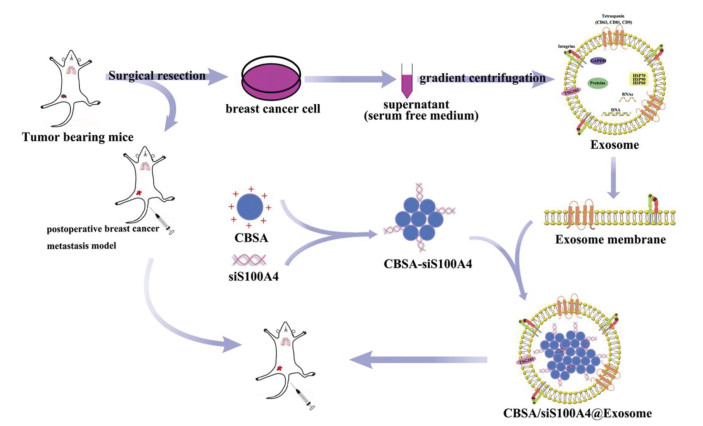
The application of tumor cell-derived exosomes was significantly influenced by cell-type tropism. For example, autologous murine colorectal cancer cells C26 derived exosomes were more efficiently taken up by C26 cells comparing with allogeneic murine melanoma cells B16BL6 derived exosomes. Even modified by PEG on these two kinds of exosomes, C26-Exo exhibited higher C26-tumor tissue accumulation and tumor-associated immune cell uptake efficiency than B16BL6 derived exosomes [95]. The influence of cell-type tropism on exosomes was utilized in different cancer types, such as pancreatic cancer, lung cancer, breast cancer and melanoma [96-99]. Actually, this interesting property of tumor cell-derived exosomes was also found in metastatic cancer, which was called organotropism. In lung metastasis model, breast cancer cell (MDA-MB-231) derived exosomes were proved to be internalized by non-small cell lung cancer cells (A549) through surfactant protein C mediated endocytosis [100]. After loading with miRNA-126, the exosomes with lung homing effect showed efficacious effect in inhibiting lung metastasis in vivo. Similarly, exosomes derived from autologous breast cancer cells loaded with cationic bovine serum albumin (CBSA) conjugated siS100A4 (CBSA/siS100A4) exhibited effective lung targeting ability in comparison to liposome loaded with CBSA/siS100A4 in 4T1-lung metastasis model as shown in Fig. 4 [96]. Another research on natural membrane-coated poly(lactic-co-glycolic acid) (PLGA) NPs also proved that tumor cell-derived exosomes consisting of both endosomal and plasma membrane proteins showed higher affinity and more superior targeting ability compared to cancer cell membrane- and lipid-coated PLGA NPs [98].
Figure 4
 Figure 4. Scheme illustration of exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. Copied with permission [96]. Copyright 2020, Elsevier.
Figure 4. Scheme illustration of exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. Copied with permission [96]. Copyright 2020, Elsevier.Besides specific cell targeting ability, autologous tumor cell-derived exosomes were also considered as specific tumor-antigen to elicit tumor-specific cytotoxic T lymphocytes for cancer immunotherapy due to immune-regulating proteins, co-stimulatory molecules and adhesion molecules contained [101, 102]. Tumor cells treated with mitoxantrone (MTX) could express immunostimulatory signals and be able to induce DC maturation [103]. MTX-treated pancreatic cancer cell-derived exosomes loaded with siCCL22 also expressed these signals and could stimulate immune responses in PDAC treatment [104]. Comparing with traditional adjuvants, exosomes seemed to be more effective with less toxic. 4T1/Her2 cell-derived exosomes loading with two immune adjuvant CpG ODN and p(I:C) displayed boosted immunostimulatory properties by activating primary and memory T cells responses and eliciting robust Th1-based immunity in 4T1 tumor bearing mice [105]. Taken together, tumor cell-derived exosomes can serve as both tumor-targeting drug carriers and tumor antigens, thus becoming novel candidates for cancer therapy. The exosomes derived from cancer patients' sera may provide opportunities for personalized immunotherapy. However, tumor-derived exosomes are considered to be a double-edged sword due to their biological functions as both antigens in immunotherapy and inducers in cancer progression. To avoid the tumor-promotion function of tumor-derived exosomes, the screening and purification of exosomes through their surface proteins are important. Paramagnetic particle method could be applied to isolate exosomes with specific protein, but the yield is low and the biological function of isolated exosome might be influenced by pH value and salt concentration [106], which would be overcome by combining the paramagnetic particle method and other methods. In addition, the genetic engineering of the tumor cells to obtain the tumor-suppression exosomes could be also an excellent alternative method.
5. Conclusions
In this review, exosomes derived from different cell sources were summarized and these exosomes could be used for different purpose depending on their contents and surface proteins. The characteristics of exosomes from different cell sources were listed in Table 3 to make it clear for selection of proper parent cells.
Table 3
Exosomes have been explored as drug carriers for delivery of different kinds of therapeutic agents. Even some exosome-based delivery systems have been applied in clinical trials, the clinical translation of exosomes still remains challenges in scale-up and manufacturing, including separation and characterization, drug loading, quality control and safety [107]. Because exosomes are a subset of EVs, the development of exosomes should follow the minimal information for studies of extracellular vesicles (MISEV) guidelines which define the parameters need to be determined such as size, morphology, and surface markers [108]. This guideline could provide some basic rules for characterization of exosomes for researchers. But for the scale-up, there is still lack of the standard operating procedure for characterization of exosomes. For different type of drugs, proper drug loading method is the key. For example, biologically accessible drugs such as nucleic acid and protein drugs could not be loaded by endogenous methods directly [107]. The genetic engineering of parent cells could be applied but is more complex in production upscaling than isolation of exosomes. The new drug loading method could pave the way for efficient loading of these molecules without complicated process. In addition, the evaluation in clinically relevant systems and the novel analytic techniques are expected to inspire the engineering of exosome-based delivery systems. As exosome is similar to the liposome in structures, the techniques in liposome development could be served as a reference. Regarding safety, the cell source is an important factor. If the large-scale production problems could be solved, autologous cells are the best choice as donors for exosomes, which seems to present lower risks than the exosomes derived from cell lines [107].
It seems that exosomes are more compliable as natural drug delivery systems, but it still needs more investigations on the side effect in normal tissues and the neoantigen effect in cancer immunotherapy. Some modification methods have been applied to enhance the targeting ability of exosomes to specific cells, meanwhile it will bring more problems for scale up such as quality control. If the source cells could be engineered to generate exosomes with specific targeting ligand and the isolation procedure could be more precise, the exosomes will be more biocompatible and easier for scale up. The influence of cell source on the surface expression and the function of exosomes remains to be investigated, which might be the principle for isolation of purified exosomes. Because cancer is a heterogenetic disease, exosome-based delivery systems have the possibility to be developed as personalized therapeutic strategies based on the understanding of the role exosomes play in cell-to-cell communication in tumor microenvironment. As efficient and safe drug delivery systems, drug loading amount in exosomes should be controllable, especially for two or more therapeutic agents co-loaded, the proper ratio is required. Exosome-based drug delivery systems will be a promising strategy in cancer therapy.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors gratefully acknowledge the support of this research by National Natural Science Foundation of China (Nos. 81971737, 32171313), Guangdong Basic and Applied Basic Research Foundation (No. 2020B1515020017), Shenzhen Science and Technology Program (No. RCYX2021070609210433), Guangdong Special Support Program (No. 2019TQ05Y224).
-
-
[1]
R. Kalluri, V.S. LeBleu, Science 367 (2020) eaau6977. doi: 10.1126/science.aau6977
-
[2]
D.M. Pegtel, S.J. Gould, Annu. Rev. Biochem. 88 (2019) 487–514. doi: 10.1146/annurev-biochem-013118-111902
-
[3]
S. Keerthikumar, D. Chisanga, D. Ariyaratne, et al., J. Mol. Biol. 428 (2016) 688–692. doi: 10.1016/j.jmb.2015.09.019
-
[4]
M. Pathan, P. Fonseka, S.V. Chitti, et al., Nucleic Acids Res. 47 (2019) D516–D519. doi: 10.1093/nar/gky1029
-
[5]
G. Arienti, E. Carlini, R. Verdacchi, et al., Biochim. Biophys. Acta 1336 (1997) 533–538. doi: 10.1016/S0304-4165(97)00071-8
-
[6]
L.M. Doyle, M.Z. Wang, Cells 8 (2019) 727. doi: 10.3390/cells8070727
-
[7]
D. Yang, W. Zhang, H. Zhang, et al., Theranostics 10 (2020) 3684–3707. doi: 10.7150/thno.41580
-
[8]
Z. Fang, X. Zhang, H. Huang, J. Wu, Chin. Chem. Lett. 33 (2022) 1693–1704. doi: 10.1016/j.cclet.2021.11.050
-
[9]
Y. Lu, Z. Tong, Z. Wu, et al., Chin. Chem. Lett. 33 (2022) 3188–3192. doi: 10.1016/j.cclet.2021.12.045
-
[10]
J. Wang, D. Chen, E.A. Ho, J. Control. Release 329 (2021) 894–906. doi: 10.1016/j.jconrel.2020.10.020
-
[11]
Y. Zhang, J. Bi, J. Huang, et al., Int. J. Nanomed. 15 (2020) 6917–6934. doi: 10.2147/IJN.S264498
-
[12]
M.N. Huda, M. Nurunnabi, Pharm. Res. 39 (2022) 2635–2671. doi: 10.1007/s11095-021-03143-4
-
[13]
P. Gao, X. Li, X. Du, et al., Front. Aging Neurosci. 13 (2021) 790863. doi: 10.3389/fnagi.2021.790863
-
[14]
Z.G. Zhang, B. Buller, M. Chopp, Nat. Rev. Neurol. 15 (2019) 193–203. doi: 10.1038/s41582-018-0126-4
-
[15]
R.C. de Abreu, H. Fernandes, P.A. da Costa Martins, et al., Nat. Rev. Cardiol. 17 (2020) 685–697. doi: 10.1038/s41569-020-0389-5
-
[16]
C.H. Chang, S. Pauklin, Cell Death Dis. 12 (2021) 973. doi: 10.1038/s41419-021-04258-7
-
[17]
L. Zhang, D. Yu, Biochim. Biophys. Acta 1871 (2019) 455–468.
-
[18]
V.C. Kok, C.C. Yu, Int. J. Nanomed. 15 (2020) 8019–8036. doi: 10.2147/IJN.S272378
-
[19]
L. Cheng, A.F. Hill, Nat. Rev. Drug. Discov. 21 (2022) 379–399. doi: 10.1038/s41573-022-00410-w
-
[20]
I. Parolini, C. Federici, C. Raggi, et al., J. Biol. Chem. 284 (2009) 34211–34222. doi: 10.1074/jbc.M109.041152
-
[21]
W. Liao, Y. Du, C. Zhang, et al., Acta Biomater. 86 (2019) 1–14. doi: 10.1016/j.actbio.2018.12.045
-
[22]
O.M. Elsharkasy, J.Z. Nordin, D.W. Hagey, et al., Adv. Drug Deliv. Rev. 159 (2020) 332–343. doi: 10.1016/j.addr.2020.04.004
-
[23]
M.S. Kim, M.J. Haney, Y. Zhao, et al., Nanomedicine 12 (2016) 655–664. doi: 10.1016/j.nano.2015.10.012
-
[24]
E.V. Batrakova, M.S. Kim, J. Control. Release 219 (2015) 396–405. doi: 10.1016/j.jconrel.2015.07.030
-
[25]
K. Maisel, M.S. Sasso, L. Potin, M.A. Swartz, Adv. Drug Deliv. Rev. 114 (2017) 43–59. doi: 10.1016/j.addr.2017.07.005
-
[26]
S. Srinivasan, F.O. Vannberg, J.B. Dixon, Sci. Rep. 6 (2016) 24436. doi: 10.1038/srep24436
-
[27]
K.B. Johnsen, J.M. Gudbergsson, M.N. Skov, et al., Biochim. Biophys. Acta 1846 (2014) 75–87.
-
[28]
S.J. Greco, P. Rameshwar, Ther. Deliv. 3 (2012) 997–1004. doi: 10.4155/tde.12.69
-
[29]
R.W. Yeo, R.C. Lai, B. Zhang, et al., Adv. Drug Deliv. Rev. 65 (2013) 336–341. doi: 10.1016/j.addr.2012.07.001
-
[30]
Z. Naseri, R.K. Oskuee, M.R. Jaafari, M. Forouzandeh Moghadam, Int. J. Nanomed. 13 (2018) 7727–7747. doi: 10.2147/IJN.S182384
-
[31]
J. Wang, G. Li, C. Tu, et al., Nanoscale 12 (2020) 13742–13756. doi: 10.1039/D0NR02344B
-
[32]
M. Zhuang, X. Chen, D. Du, et al., Nanoscale 12 (2020) 173–188. doi: 10.1039/C9NR05865F
-
[33]
Z. Yang, J. Shi, J. Xie, et al., Nat. Biomed. Eng. 4 (2020) 69–83.
-
[34]
A. Kabashima-Niibe, H. Higuchi, H. Takaishi, et al., Cancer Sci. 104 (2013) 157–164. doi: 10.1111/cas.12059
-
[35]
W. Blogowski, T. Bodnarczuk, T. Starzynska, Stem. Cells Transl. Med. 5 (2016) 938–945. doi: 10.5966/sctm.2015-0291
-
[36]
Y. Zhou, W. Zhou, X. Chen, et al., Acta Pharm. Sin. B 10 (2020) 1563–1575. doi: 10.1016/j.apsb.2019.11.013
-
[37]
W. Zhou, Y. Zhou, X. Chen, et al., Biomaterials 268 (2021) 120546. doi: 10.1016/j.biomaterials.2020.120546
-
[38]
G. Lou, L. Chen, C. Xia, et al., J. Exp. Clin. Cancer Res. 39 (2020) 4. doi: 10.1186/s13046-019-1512-5
-
[39]
A. Thakur, R.K. Sidu, H. Zou, et al., Int. J. Nanomed. 15 (2020) 8331–8343. doi: 10.2147/IJN.S263956
-
[40]
T. Vilanova-Perez, C. Jones, S. Balint, et al., Nanomedicine (Lond) 15 (2020) 1965–1980. doi: 10.2217/nnm-2020-0056
-
[41]
X. Zhu, M. Badawi, S. Pomeroy, et al., J. Extracell. Vesicles 6 (2017) 1324730. doi: 10.1080/20013078.2017.1324730
-
[42]
L.E. Rosas, O.A. Elgamal, X. Mo, et al., J. Immunotoxicol. 13 (2016) 652–665. doi: 10.3109/1547691X.2016.1148089
-
[43]
Y. Si, S. Kim, E. Zhang, et al., Biotechnol. J. 15 (2020) e1900163. doi: 10.1002/biot.201900163
-
[44]
A. Afshari, C. Uhde-Stone, B. Lu, Biochem. Biophys. Res. Commun. 448 (2014) 281–286. doi: 10.1016/j.bbrc.2014.04.108
-
[45]
N. Duong, K. Curley, A. Brown, et al., Int. J. Nanomed. 14 (2019) 3413–3425. doi: 10.2147/IJN.S196975
-
[46]
G. Liang, Y. Zhu, D.J. Ali, et al., J. Nanobiotechnol. 18 (2020) 10. doi: 10.1186/s12951-019-0563-2
-
[47]
E. Cho, G.H. Nam, Y. Hong, et al., J. Control. Release 279 (2018) 326–335. doi: 10.1016/j.jconrel.2018.04.037
-
[48]
N. Yim, S.W. Ryu, K. Choi, et al., Nat. Commun. 7 (2016) 12277. doi: 10.1038/ncomms12277
-
[49]
H. Choi, Y. Kim, A. Mirzaaghasi, et al., Sci. Adv. 6 (2020) eaaz6980. doi: 10.1126/sciadv.aaz6980
-
[50]
R.E. Veerman, G. Gucluler Akpinar, M. Eldh, S. Gabrielsson, Trends Mol. Med. 25 (2019) 382–394. doi: 10.1016/j.molmed.2019.02.003
-
[51]
T. Lawrence, G. Natoli, Nat. Rev. Immunol. 11 (2011) 750–761. doi: 10.1038/nri3088
-
[52]
A. Mantovani, S. Sozzani, M. Locati, et al., Trends Immunol. 23 (2002) 549–555. doi: 10.1016/S1471-4906(02)02302-5
-
[53]
S.K. Biswas, A. Mantovani, Nat. Immunol. 11 (2010) 889–896. doi: 10.1038/ni.1937
-
[54]
C.X. Li, Y. Zhang, X. Dong, et al., Adv. Mater. 31 (2019) e1807211. doi: 10.1002/adma.201807211
-
[55]
L. Cheng, Y. Wang, L. Huang, Mol. Ther. 25 (2017) 1665–1675. doi: 10.1016/j.ymthe.2017.02.007
-
[56]
P.P. Wang, H.H. Wang, Q.Q. Huang, et al., Theranostics 9 (2019) 1714–1727. doi: 10.7150/thno.30716
-
[57]
G.R. Gunassekaran, S.M. Poongkavithai Vadevoo, M.C. Baek, B. Lee, Biomaterials 278 (2021) 121137. doi: 10.1016/j.biomaterials.2021.121137
-
[58]
M.J. Haney, N.L. Klyachko, Y.L. Zhao, et al., J. Control. Release 207 (2015) 18–30. doi: 10.1016/j.jconrel.2015.03.033
-
[59]
Y. Binenbaum, E. Fridman, Z. Yaari, et al., Cancer Res. 78 (2018) 5287–5299.
-
[60]
E. Behzadi, H.M. Hosseini, R. Halabian, A.A.I. Fooladi, Microb. Pathog. 111 (2017) 132–138. doi: 10.1016/j.micpath.2017.08.027
-
[61]
C. Gong, J. Tian, Z. Wang, et al., J. Nanobiotechnol. 17 (2019) 93. doi: 10.1186/s12951-019-0526-7
-
[62]
W. Nie, G. Wu, J. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 2018–2022. doi: 10.1002/anie.201912524
-
[63]
M.S. Kim, M.J. Haney, Y.L. Zhao, et al., Nanomed. Nanotechnol. Biol. Med. 14 (2018) 195–204. doi: 10.1016/j.nano.2017.09.011
-
[64]
G. Jia, Y. Han, Y.L. An, et al., Biomaterials 178 (2018) 302–316. doi: 10.1016/j.biomaterials.2018.06.029
-
[65]
S. Li, Y. Wu, F. Ding, et al., Nanoscale 12 (2020) 10854–10862. doi: 10.1039/D0NR00523A
-
[66]
S. Rayamajhi, T.D.T. Nguyen, R. Marasini, S. Aryal, Acta Biomater. 94 (2019) 482–494. doi: 10.1016/j.actbio.2019.05.054
-
[67]
S.K. Wculek, F.J. Cueto, A.M. Mujal, et al., Nat. Rev. Immunol. 20 (2020) 7–24. doi: 10.1038/s41577-019-0210-z
-
[68]
N.L. Vujanovic, Immunol. Res. 50 (2011) 159–174. doi: 10.1007/s12026-011-8228-8
-
[69]
G. Lu, B.M. Janjic, J. Janjic, et al., J. Immunol. 168 (2002) 1831–1839. doi: 10.4049/jimmunol.168.4.1831
-
[70]
I. Marigo, S. Zilio, G. Desantis, et al., Cancer Cell 30 (2016) 377–390. doi: 10.1016/j.ccell.2016.08.004
-
[71]
S. Munich, A. Sobo-Vujanovic, W.J. Buchser, et al., Oncoimmunology 1 (2012) 1074–1083. doi: 10.4161/onci.20897
-
[72]
Z. Lu, B. Zuo, R. Jing, et al., J. Hepatol. 67 (2017) 739–748. doi: 10.1016/j.jhep.2017.05.019
-
[73]
J.M. Pitt, M. Charrier, S. Viaud, et al., J. Immunol. 193 (2014) 1006–1011. doi: 10.4049/jimmunol.1400703
-
[74]
L. Zitvogel, A. Regnault, A. Lozier, et al., Nat. Med. 4 (1998) 594–600. doi: 10.1038/nm0598-594
-
[75]
F. Andre, N. Chaput, N.E. Schartz, et al., J. Immunol. 172 (2004) 2126–2136. doi: 10.4049/jimmunol.172.4.2126
-
[76]
B. Escudier, T. Dorval, N. Chaput, et al., J. Transl. Med. 3 (2005) 10. doi: 10.1186/1479-5876-3-10
-
[77]
M. Xu, Q. Chen, J. Li, et al., J. B.U. ON. 25 (2020) 1413–1422.
-
[78]
L. Alvarez-Erviti, Y. Seow, H. Yin, et al., Nat. Biotechnol. 29 (2011) 341–345. doi: 10.1038/nbt.1807
-
[79]
Y. Tian, S. Li, J. Song, et al., Biomaterials 35 (2014) 2383–2390. doi: 10.1016/j.biomaterials.2013.11.083
-
[80]
L. Xin, Y.W. Yuan, C. Liu, et al., Dig. Dis. Sci. 66 (2021) 1045–1053. doi: 10.1007/s10620-020-06262-x
-
[81]
L. Chiossone, P.Y. Dumas, M. Vienne, E. Vivier, Nat. Rev. Immunol. 18 (2018) 671–688. doi: 10.1038/s41577-018-0061-z
-
[82]
L. Zhu, J.M. Oh, P. Gangadaran, et al., Front. Immunol. 9 (2018) 824. doi: 10.3389/fimmu.2018.00824
-
[83]
L. Zhu, S. Kalimuthu, P. Gangadaran, et al., Theranostics 7 (2017) 2732–2745. doi: 10.7150/thno.18752
-
[84]
L. Lugini, S. Cecchetti, V. Huber, et al., J. Immunol. 189 (2012) 2833–2842. doi: 10.4049/jimmunol.1101988
-
[85]
A.Y. Jong, C.H. Wu, J. Li, et al., J. Extracell. Vesicles 6 (2017) 1294368. doi: 10.1080/20013078.2017.1294368
-
[86]
D. Han, K. Wang, T. Zhang, et al., Eur. Rev. Med. Pharmacol. Sci. 24 (2020) 5703–5713.
-
[87]
P. Neviani, P.M. Wise, M. Murtadha, et al., Cancer Res. 79 (2019) 1151–1164.
-
[88]
G. Wang, W. Hu, H. Chen, et al., Cancers 11 (2019) 1560. doi: 10.3390/cancers11101560
-
[89]
R. Xu, A. Rai, M. Chen, et al., Nat. Rev. Clin. Oncol. 15 (2018) 617–638. doi: 10.1038/s41571-018-0036-9
-
[90]
Y. Tan, X. Luo, W. Lv, et al., Cell Death Dis. 12 (2021) 547. doi: 10.1038/s41419-021-03825-2
-
[91]
P.H.L. Tran, T. Wang, W. Yin, et al., Int. J. Pharm. 572 (2019) 118786. doi: 10.1016/j.ijpharm.2019.118786
-
[92]
Y. Jang, H. Kim, S. Yoon, et al., J. Control. Release 330 (2021) 293–304. doi: 10.1016/j.jconrel.2020.12.039
-
[93]
M. Morishita, Y. Takahashi, M. Nishikawa, et al., Mol. Pharm. 14 (2017) 4079–4086. doi: 10.1021/acs.molpharmaceut.7b00760
-
[94]
W. Zhang, F. Wang, C. Hu, et al., Acta Pharm. Sin. B 10 (2020) 2037–2053. doi: 10.1016/j.apsb.2020.07.013
-
[95]
S.E. Emam, A.S. Abu Lila, N.E. Elsadek, et al., Eur. J. Pharm. Biopharm. 145 (2019) 27–34. doi: 10.1016/j.ejpb.2019.10.005
-
[96]
L. Zhao, C. Gu, Y. Gan, et al., J. Control. Release 318 (2020) 1–15. doi: 10.1016/j.jconrel.2019.12.005
-
[97]
L. Xu, F.N. Faruqu, Y.M. Lim, et al., Biomaterials 264 (2021) 120369. doi: 10.1016/j.biomaterials.2020.120369
-
[98]
C. Liu, W. Zhang, Y. Li, et al., Nano Lett. 19 (2019) 7836–7844. doi: 10.1021/acs.nanolett.9b02841
-
[99]
T. Yong, X. Zhang, N. Bie, et al., Nat. Commun. 10 (2019) 3838. doi: 10.1038/s41467-019-11718-4
-
[100]
H. Nie, X. Xie, D. Zhang, et al., Nanoscale 12 (2020) 877–887. doi: 10.1039/C9NR09011H
-
[101]
J. Wolfers, A. Lozier, G. Raposo, et al., Nat. Med. 7 (2001) 297–303. doi: 10.1038/85438
-
[102]
T.H. Tran, G. Mattheolabakis, H. Aldawsari, M. Amiji, Clin. Immunol. 160 (2015) 46–58. doi: 10.1016/j.clim.2015.03.021
-
[103]
M. Obeid, A. Tesniere, F. Ghiringhelli, et al., Nat. Med. 13 (2007) 54–61. doi: 10.1038/nm1523
-
[104]
W. Zhou, X. Chen, Y. Zhou, et al., Biomaterials 280 (2022) 121306. doi: 10.1016/j.biomaterials.2021.121306
-
[105]
M. Yildirim, T.C. Yildirim, N. Turay, et al., Immunol. Lett. 239 (2021) 32–41. doi: 10.1016/j.imlet.2021.08.004
-
[106]
C. Admyre, S.M. Johansson, K.R. Qazi, et al., J. Immunol. 179 (2007) 1969–1978. doi: 10.4049/jimmunol.179.3.1969
-
[107]
I.K. Herrmann, M.J.A. Wood, G. Fuhrmann, Nat Nanotechnol. 16 (2021) 748–759. doi: 10.1038/s41565-021-00931-2
-
[108]
C. Thery, K.W. Witwer, E. Aikawa, et al., J. Extracell. Vesicles 7 (2018) 1535750. doi: 10.1080/20013078.2018.1535750
-
[1]
-
Figure 1 Scheme of exosomes derived from different sources as drug delivery systems for loading small molecules, nucleic acids and proteins in cancer therapy. MSCs, mesenchymal stem cells; DCs, dendritic cells; NKs, natural killer cells; PANC, pancreatic cell.
Figure 2 Schematic of OXA and galectin-9 siRNA co-loaded delivery system for pancreatic cancer treatment through enhancing immune response and reprogramming tumor microenvironment. Copied with permission [37]. Copyright 2021, Elsevier.
Figure 3 Schematic of Mn2+-induced M1 macrophage polarization and the synergistic anticancer effect of M1-Exo engineered with aCD47 and aSIRPα. Copied with permission [62]. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 4 Scheme illustration of exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. Copied with permission [96]. Copyright 2020, Elsevier.
-

 DownLoad:
DownLoad:




 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 29
- 文章访问数: 3052
- HTML全文浏览量: 378
