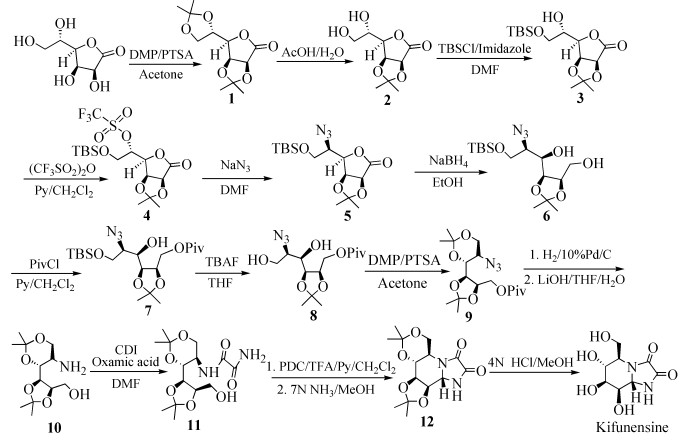
图式 1.
Kifunensine的改进合成路线
Scheme 1.
Improved synthetic route for Kifunensine

Kifunensine是甘露糖苷酶Ⅰ的有效抑制剂[1, 2],它于1987年由Iwami等从名为Kitasatosporia kifunense的放线菌中分离纯化得到[3],是一种中性、稳定的生物碱[4]。Kifunensine作为一类中性分子,与其他甘露糖苷酶抑制剂相比,它可以迅速渗透到细胞内,在细胞内它可以防止内质网甘露糖苷酶Ⅰ修复前体糖蛋白中的甘露糖残基,从而起到抑制糖基化的作用,同时它对甘露糖苷酶Ⅱ没有抑制作用,对芳基甘露糖苷酶的抑制作用也很弱。因此,当将其掺入细胞培养基中时,Kifunensine对细胞生长或糖蛋白产量不会有显著影响,但可以大大提高所制备糖蛋白的品质[5]。研究发现,Kifunensine还具有抗HIV活性,并且由于具有免疫调节作用,被用于抗肿瘤药物的开发[6];此外,Kifunensine已经于2011年被欧洲药品管理局批准为用于治疗肢体-肌束营养不良症的单独用药。同时研究发现,Kifunensine可以在极低的浓度下抑制内质网甘露糖苷酶Ⅰ的活性并干扰早期基质的识别,从而抑制内质网的自身降解,进而可以用于治疗溶酶体存储障碍的相关疾病,如台-萨氏综合症。因此,Kifunensine具有很高的应用价值。
1989年,Kifunensine的具体分子结构由Kayakiri等鉴定得到,并于次年由他们人工合成[7]。但是它的合成制备非常困难,存在收率低、合成成本高等问题,市场售价十分昂贵。目前对于Kifunensine的合成报道相对较少,有效合成路线仅两条:一条是以N-乙酰基-D-甘露糖胺为起始原料经过羟基保护、脱保护、酰化、氧化关环等8步反应转得到[7],该路线的可重复性较差、收率低且原料价格较为昂贵;另一条是以L-抗坏血酸为起始原料经过15步反应得到[8],虽然该方法的原料价格较为便宜,但是该方法第一步氢气还原反应时间较长,且需要加压,导致反应收率低、可重复性差以及产物不易分离等问题,此外该方法制备过程中还用到了叠氮酸等易爆试剂,不利于进一步放大。
鉴于Kifunensine潜在的应用价值,本文在前期研究的基础上,重新设计了一条它的改进合成路线(图式 1)。该方法以廉价易得的L-古洛糖酸-γ-内酯为起始原料,先对糖的2、3、5、6位羟基进行缩酮保护,随后产物不经纯化直接用乙酸水溶液处理得到5、6位羟基去保护的糖中间体2。不经纯化继续对糖的6位羟基进行硅醚保护,随后对剩余的5位羟基进行三氟甲基磺酰化得到中间体4;之后中间体4在室温下用叠氮化钠处理得到叠氮糖中间体5。中间体5用硼氢化钠还原得到糖环打开的中间体6,之后用特戊酰基保护1位羟基得到中间体7;随后用四丁基氟化铵处理脱去6位羟基的硅醚保护得到中间体8;之后再次对剩余的4、6位羟基进行缩酮保护得到中间体9。中间体9随后进行催化加氢反应将叠氮基团还原为氨基,之后不经纯化继续用氢氧化锂水解酯基得到中间体10,之后在草胺酸的作用下反应得到中间体11;随后对1位羟基进行氧化得到醛基中间体,适当纯化后用氨气的甲醇溶液处理关环得到氮杂糖12,最后用盐酸的甲醇溶液处理得到Kifunensine。反应总收率为4.8%。操作中的多个步骤可以用一锅法投料,较为简便。该方法为Kifunensine的大规模制备提供了新的思路。
Bruker Plus 400M型核磁共振谱仪,Waters 3100 Mass Detctor质谱仪。实验所用柱层析硅胶(200~300目)及薄层层析硅胶板购自青岛海洋化工厂。其他试剂均为国产或进口分析纯级,无水溶剂用常规方法干燥处理。
(3aS, 6R, 6aS)-6-((S)-2, 2-二甲基-1, 3-二氧戊烷-4-基)-2, 2-二甲基二氢呋喃[3, 4-d][1, 3]二氧-4(3aH)-酮(1)的合成:将20g(112.3mmol) L-古洛糖酸-γ-内酯和1.9g(11.2mmol)对甲苯磺酸(PTSA)加入到300mL丙酮中搅拌,之后加入55.2mL(449.1mmol) 2, 2-二甲氧基丙烷(DMP),氩气保护,室温反应,体系由白色悬浊液变为黄色澄清液,反应过程中及时用真空泵减压除去产生的甲醇,6h后TLC监测原料反应完毕,之后加入饱和碳酸氢钠溶液调节体系pH至中性,旋蒸除去溶剂,加二氯甲烷稀释剩余油状物,用饱和氯化钠洗涤3次,收集有机相,加入无水Na2SO4干燥,过滤,旋蒸除去溶剂得23.6g淡黄色固体,即中间体1,不经纯化直接用于下一步反应。
(3aS, 6R, 6aS)-6-((S)-1, 2-二羟基乙基)-2, 2-二甲基二氢呋喃[3, 4-d][1, 3]二氧杂环-4(3aH)-酮(2)的合成:将上一步得到的23.6g中间体1置于320mL 80%的乙酸水溶液中搅拌,体系为无色澄清液,之后在氩气保护下,室温反应至原料转化完全,减压浓缩除去溶剂,得到15g白色固体,即中间体2,不经纯化直接用于下一步反应。
(3aS, 6R, 6aS)-6-((S)-2-((叔丁基二甲基硅烷基)氧基)-1-羟乙基)-2, 2-二甲基二氢呋喃[3, 4-d] [1, 3]二氧杂环-4(3aH)-酮(3)的合成:将15g中间体2和7g(103mmol)咪唑加入到100mL无水DMF中搅拌,氩气保护并冷却至-40℃,之后缓慢向反应体系中滴入16.6g(110mmol)叔丁基二甲基氯硅烷(TBSCl)的50mL DMF溶液,滴加结束后继续搅拌反应2.5h,TLC监测至原料反应完毕,加水淬灭反应,旋蒸除去溶剂,加入二氯甲烷稀释剩余油状物,用饱和氯化钠溶液洗涤3次,之后加入无水硫酸钠干燥,过滤,减压浓缩除去溶剂,经硅胶柱层析纯化得15.5g无色油状物,即中间体3,三步反应总收率为42%。1H NMR (600MHz,CDCl3) δ:4.88~4.81(m,2H),4.57(m,1H),4.07(m,1H),3.86~3.78(m,2H),1.49(s,3H),1.40(s,3H),0.91(s,9H),0.10(d,J=2.3Hz,6H);ESI-MS m/z:理论值C15H29O6Si [M+H]+ 333.46,实测值333.36。
(S)-2-((叔丁基二甲基硅烷基)氧基)-1-((3aS, 4S, 6aS)-2, 2-二甲基-6-氧四氢呋喃[3, 4-d][1, 3]二氧杂环-4-基)三氟甲磺酸乙酯(4)的合成:称取5.5g(16.5mmol)化合物3溶于60mL二氯甲烷中,体系冷却至-30℃,之后加入4mL(49.6mmol)吡啶以及5.4mL(33.1mmol)三氟甲烷磺酸酐,体系变为橙色澄清液,继续搅拌至原料反应完毕。之后,加入大量二氯甲烷,用2mol/L盐酸洗涤一次,分离有机相,之后用饱和碳酸氢钠溶液洗涤一次,加入无水硫酸钠干燥,过滤,旋蒸除去溶剂得6.1g橙色固体,即中间体4,不经纯化直接用于下一步反应。
(3aS, 6R, 6aS)-6-((R)-1-叠氮基-2-((叔丁基二甲基硅烷基)氧基)乙基)-2, 2-二甲基二氢呋喃[3, 4-d][1, 3]二氧杂环-4(3aH)-酮(5)的合成:室温条件下将6.1g(13.1mmol)中间体4溶于55mL DMF中,之后加入2.6g(39.4mmol)叠氮化钠,体系变为淡黄色悬浊液,室温搅拌至原料反应完毕,之后,加水淬灭反应,向体系中加入适量二氯甲烷,之后用饱和氯化钠溶液洗涤3次,收集有机相,加入无水硫酸钠干燥,过滤,减压浓缩除去溶剂,硅胶柱层析得4.3g黄色油物,即中间体5,两步反应总收率73%。1H NMR (400MHz,CDCl3)δ:4.93~4.79(m,2H),4.39(m,1H),4.06(m,1H),3.87(m,1H),3.76(m,1H),1.49(s,3H),1.44(s,3H),0.92(s,9H),0.11(d,J=1.5Hz,6H)。
(1R, 2R)-2-叠氮基-3-((叔丁基二甲基硅基)氧基)-1-((4S, 5R)-5-(羟甲基)-2, 2-二甲基-1, 3-二氧戊环-4-基)丙-1-醇(6)的合成:称取2.3g(6.4mmol)中间体5溶于50mL无水乙醇中,体系呈淡黄色澄清液,之后加入487mg(12.9mmol)硼氢化钠,过程中体系有少量气泡产生,室温反应过夜,反应完毕后加水淬灭,减压浓缩除去乙醇,加入二氯甲烷稀释剩余油状物,用饱和氯化钠溶液洗涤3次,收集有机相,加入无水硫酸钠干燥,过滤,减压浓缩除去溶剂得2g无色油状物,即中间体6,收率86%。1H NMR (600MHz,CDCl3)δ:4.41(m,1H),4.30 (m,1H),4.09 (m,1H),3.90~3.85 (m,2H),3.80 (m,1H),3.62 (m,1H),3.49 (m,1H),1.53 (s,3H),1.41 (s,3H),0.92 (s,9H),0.10 (d,J=3.2 Hz,6H);ESI-MS m/z:C15H31N3O5SiNa [M+Na]+,理论值384.51,实测值384.42。
((4R, 5S)-5-((1R, 2R)-2-叠氮基-3-((叔丁基二甲基硅基)氧基)-1-羟丙基)-2, 2-二甲基-1, 3-二氧戊环-4-基)甲基新戊酸酯(7)的合成:将上一步得到的2g (5.5mmol)中间体6溶于20mL二氯甲烷中,加入1.4mL(17mmol)吡啶以及1mL(8.3mmol)特戊酰氯(PivCl),室温搅拌反应过夜,TLC监测至原料反应完毕后,加入适量二氯甲烷稀释,用饱和氯化钠溶液洗涤3次,收集有机相,加入无水硫酸钠干燥,过滤,浓缩后经硅胶柱层析纯化得2.3g无色油状物,即中间体7,收率93%。1H NMR (600MHz,CDCl3) δ:4.44~4.38 (m,2H),4.34(m,2H),4.08(m,1H),3.87(m,1H),3.60 (m,1H),3.39 (m,1H),2.37(d,J=9.1Hz,1H),1.50(s,3H),1.39(s,3H),1.21(s,9H),0.91(s,9H),0.10(d,J=4.0Hz,6H)。
((4R, 5S)-5-((1R, 2R)-2-叠氮基-1, 3-二羟基丙基-2, 2-二甲基-1, 3-二氧戊烷-4-基)甲基新戊酸酯(8)的合成:将上一步得到的2.3g(5.2mmol)化合物7加入到30mL四氢呋喃中,之后将体系冷却至-20℃,加入6mL 1mol/L的四丁基氟化铵的THF溶液,继续搅拌反应2h,TLC监测至原料反应完毕,加入少量饱和氯化钠溶液搅拌,之后用乙酸乙酯萃取,收集有机相,加入无水硫酸钠干燥,过滤,减压浓缩除去溶剂,硅胶柱层析得1.68g无色油状物,即化合物8,收率98%。1H NMR (400MHz,CDCl3)δ:4.42(m,2H),4.36~4.31 (m,2H),4.11(m,1H),4.01(m,1H),3.89 (m,1H),3.69(m,1H),3.52~3.46(m,1H),2.46(d,J=9.1Hz,1H),1.51(s,3H),1.40(s,3H),1.21(s,9H);ESI-MS m/z:C14H25N3O6Na[M+Na]+,理论值354.36,实测值354.34。
((4R, 5S)-5-((4R, 5S)-5-叠氮基-2, 2-二甲基-1, 3-二噁烷-4-基)-2, 2-二甲基-1, 3-二氧戊环-4-基)甲基新戊酸酯(9)的合成:将1.68g(5.1mmol)化合物8与87mg(0.51mmol)PTSA溶于20mL丙酮中,加入2.5mL(20.3mmol)DMP,室温条件下搅拌反应,反应过程中用真空泵减压除去产生的甲醇,体系搅拌至TLC监测原料反应完毕为止,之后加入饱和碳酸氢钠溶液调节体系pH至中性,旋蒸除去溶剂,加二氯甲烷稀释剩余油状物,用饱和氯化钠洗涤3次,收集有机相,加入无水硫酸钠干燥,过滤,减压浓缩除去溶剂,经硅胶柱层析纯化得1.5g淡黄色固体,即中间体9,产率为80%。1H NMR (400MHz,CDCl3)δ:4.51~4.40 (m,1H),4.39~4.31(m,3H),4.06~4.00(m,1H),3.83~3.70 (m,2H),3.59(m,1H),1.53(s,3H),1.46(s,3H),1.42(s,3H),1.40(s,3H),1.24(d,J=4.1Hz,9H);ESI-MS:C17H30N3O6 [M+H]+,理论值371.43,实测值371.58。
((4R, 5S)-5-((4R, 5S)-5-氨基-2, 2-二甲基-1, 3-二噁烷-4-基)-2, 2-二甲基-1, 3-二氧戊烷-4-基)甲醇(10)的合成:称取1.5g(2.2mmol)中间体9溶解于20mL甲醇中,加入150mg钯碳,气体置换后进行常压加氢反应,室温反应6h,TLC监测原料反应完,抽滤除去催化剂,并将滤液旋干,得到无色油状物,将其溶于20mL甲醇中,加入79mg(3.3mmol)氢氧化锂,并加入1mL水,体系为无色澄清,室温继续反应4h,TLC监测至原料反应完毕,抽滤除去不溶物,并将滤液旋干,剩余物经过硅胶柱纯化得到1.03g无色油状中间体10,产率98%。1H NMR (600MHz,CDCl3) δ:4.49 (m,1H),4.27 (m,1H),3.88 (m,1H),3.77 (m,2H),3.56~3.43 (m,2H),3.10 (m,1H),1.85 (s,3H),1.52 (s,3H),1.47 (s,3H),1.42 (s,3H),1.38 (s,3H);ESI-MS m/z:C12H24NO5 [M+H]+,理论值262.31,实测值262.67。
N1-((4R, 5S)-4-((4S, 5R)-5-(羟甲基)-2, 2-二甲基-1, 3-二氧戊环-4-基)-2, 2-二甲基-1, 3-二噁烷-5-基)草酰胺(11)的合成:称取180mg(2.02mmol)草氨酸,将其溶于5mL DMF中,60℃加热搅拌30min,冷却至室温,之后加入360mg(2.22mmol)N, N′-羰基二咪唑(CDI),室温搅拌1h,然后缓慢滴加0.36g(1.38mmol)中间体10的5mL DMF溶液,室温搅拌反应过夜,反应完毕后,加入1mL水继续搅拌30min,减压浓缩除去溶剂,剩余物进行硅胶柱纯化得318mg白色固体中间体11,产率69%。1H NMR (600MHz,DMSO-d6)δ:8.69(d,J=9.4Hz,1H),8.10(d,J=2.2Hz,1H),7.92~7.77(m,1H),4.85(m,1H),4.21(m,1H),4.04(m,1H),3.99~3.88 (m,2H),3.71~3.63(m,2H),3.58(m,2H),1.45(s,3H),1.41(s,3H),1.28(s,3H),1.24 (s,4H);ESI-MS m/z:C14H23N2O7 [M-H]-,理论值331.35,实测值331.37。
(3aS, 3bR, 7aS, 11aS, 11bR)-2, 2, 5, 5-四甲基四氢-3aH-[1, 3]二氧己环并[4, 5-e][1, 3]二氧戊环并[4, 5-c]咪唑并[1, 2-a]吡啶-9, 10(3bH, 11bH)-二酮(12)的合成:将318mg(0.96mmol)中间体11溶于5mL二氯甲烷中,之后依次加入720mg(1.91mmol)重铬酸吡啶盐(PDC)、71μL(0.96mmol)三氟乙酸以及76μL(0.96mmol)吡啶,氩气保护,室温搅拌反应5h,TLC监测至反应完全,过滤除去不溶物,收集滤液,减压浓缩除去溶剂得红棕色油状液体。之后向其中加入5mL 7mol/L氨气的甲醇溶液,氩气保护,室温搅拌反应过夜。反应完毕后,减压浓缩除去溶剂,加入适量饱和碳酸氢钠溶液搅拌,并加乙酸乙酯进行萃取,之后收集有机相,加入无水硫酸钠干燥,过滤,减压浓缩除去溶剂,经硅胶柱纯化得120mg白色固体中间体12,两步反应收率40%。1H NMR (600MHz,CDCl3)δ:4.86(d,J=8.6Hz,1H),4.75(dd,J=11.1、4.9 Hz,1H),4.39(t,J=8.0Hz,1H),4.22(dd,J=11.3、8.1 Hz,1H),4.00(t,J=8.3Hz,1H),3.81(t,J=10.7Hz,1H),3.63(m,1H),1.58(d,J=5.9Hz,6H),1.51(s,3H),1.39(s,3H);ESI-MS m/z:C14H19N2O6 [M-H]-,理论值311.32,实测值311.34。
Kifunensine的合成:将120mg(0.38mmol)中间体12溶于2mL 4mol/L氯化氢的甲醇溶液中,室温搅拌反应1h后,TLC监测反应完全,减压浓缩除去溶剂,用甲醇分散洗涤,最终得白色固体83mg,即Kifunensine,收率93%。1H NMR(600MHz,DMSO-d6)δ:10.38~10.05(m,1H),5.44(s,2H),4.71(m,1H),4.08(m,1H),3.86(m,1H),3.77(t,J=3.2Hz,1H),3.67(m,1H),3.53 (m,1H),3.30 (m,1H);13C NMR (151MHz,CDCl3)δ:165.42,163.43,77.25,77.07,73.99,67.72,65.11,63.44;ESI-MS m/z:C8H13N2O6 [M+H]+,理论值233.19,实测值233.28。
中间体1是L-古洛糖酸-γ-内酯羟基保护后的产物,目前已报道文献中是用浓硫酸催化[8],以丙酮为溶剂的方法进行羟基的缩酮保护,这种方法收率极低,反应需要长时间加热。本文使用2, 2-二甲氧基丙烷进行羟基保护,并在反应进行中及时减压除去甲醇,减少了开环副产物的产生,提高了反应速率,使反应在常温进行,极大地提高了产率。
中间体5为叠氮糖中间体,文献中使用叠氮酸进行反应[8],反应危险性大,不适合大量生产。本文采用对5位羟基进行三氟甲基磺酰化保护形成离去基团,再用叠氮化钠进攻取代磺酸基的方法进行叠氮化反应,反应条件较叠氮酸安全、温和且适合大批量投料。
文献在进行中间体9的合成时,采用2-甲氧基丙烯进行羟基的保护[8],这种方法反应速率虽然快,但是副产物极多,体系较乱,不易纯化。本文采用2, 2-二甲氧基丙烷的方法进行羟基保护,反应相对缓和,副产物少,易于纯化,产率高。
文献在进行中间体10的合成时,采用的是氢化铝锂还原[8],投料过程中氢化铝锂接近5倍量,体系需要低温冷却,在放大制备时具有一定危险性,且反应后处理会产生大量固废,导致后处理困难以及环境污染等问题。本文采取先用钯碳催化加氢还原氨基,再继续水解酯基的方法,反应过程中钯碳催化剂可以回收重复利用,且整体操作十分便捷,安全性高。用这种方法几乎可以定量得到中间体10,而用氢化铝锂还原的方法,收率仅为60%左右。
由于蛋白的糖基化修饰中糖链存在不均一性,使得通过酶去糖基化不能达到完全除去糖基的目的,从而导致表达纯化的蛋白均一性差,因此需要加入化学物质和酶相互配合,以除去相同类型糖蛋白的糖或糖脂的糖,从而达到提高蛋白的均一性的目的。本实验中通过Kifunensine和糖苷内切酶Endo H的相互配合使用来提高蛋白的均一性。
在内质网中前体低聚糖链经过α-葡萄糖苷酶Ⅰ、α-葡萄糖苷酶Ⅱ切割3分子葡萄糖形成高甘露糖型,随后α-甘露糖苷酶Ⅰ切割1分子甘露糖,得到中间体高尔基体,高尔基体中α-甘露糖苷酶Ⅰ继续切割甘露糖,并经过各种糖基转移酶修饰形成复杂型、杂合型的N-糖基化修饰。Kifunensine是α-甘露糖苷酶Ⅰ的抑制剂,其抑制机制是抑制内质网中α-甘露糖苷酶Ⅰ对甘露糖的切割,从而形成高甘露糖型修饰的N-糖基化,进而抑制N-糖基化的复杂化修饰,最终避免高甘露糖型形成复杂型、杂合型的N-糖基化修饰。总体而言,Kifunensine可使N-糖基化形成单一的高甘露糖型。糖苷内切酶Endo H是一种良好的去高甘露糖型糖基化的糖苷酶,主要识别两个N-乙酰葡萄糖胺之间的糖苷键,Endo H将N-糖基化的糖链进行切割,最终仅残留1个N-乙酰葡萄糖胺在氨基酸残基上面。因此Kifunensine和Endo H相互配合除去N-糖基化从而提高表达纯化的蛋白均一性的机制是:Kifunensine使N-糖基化形成单一的高甘露糖型,然后Endo H对高甘露糖型的糖链进行切割,仅残留1个N-乙酰葡萄糖胺在蛋白质氨基酸残基上,使得蛋白的均一化程度大大提升,此类方法可以用来进行表面膜蛋白的表达研究。
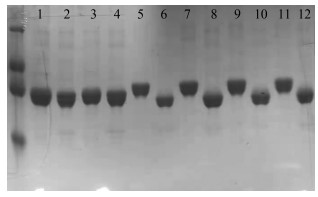
根据以上原理,对本文合成的Kifunensine与商业购买的Kifunensine对照品进行活性对比,在蛋白表达纯化过程中加入相应化合物和酶,之后进行聚丙烯酰胺凝胶电泳,其结果如图 1所示。最左侧条带为蛋白marker,其中1、3条带为不加Kifunensine的空白对照;2、4条带为空白对照用EndoH酶切;5、7条带为商业购买Kifunensine对照品;6、8条带为合成的Kifunensine对照品然后用Endo H酶切;9、11条带为Kifunensine合成品;10、12条带为Kifunensine合成品然后用Endo H酶切。由条带5~12的位置对比可见,该方法制备的Kifunensine生物活性的评价效果与商业购买的对照品一致。
本文对Kifunensine的合成路线进行了优化和改进,以L-古洛糖酸-γ-内酯为原料,经过羟基保护、叠氮化、还原开环、酰化、氧化关环、脱保护等多步反应高效地得到Kifunensine,反应总收率可达4.8%。反应操作中多个步骤可以用一锅法投料,操作简便,合成过程中避免了危险试剂的使用以及减少了环境的污染。该路线方法目前已经进行了多次实验,重现性很好;此外,通过对该方法制备的Kifunensine进行了生物活性的评价,其效果与商业购买的对照品类似。因此,本文报道的Kifunensine的改进合成方法为该化合物的大规模制备提供了新的思路。
M Iwami, O Nakayama, H Terano et al. J. Antibiot., 1987, 40(5): 612~622. doi: 10.7164/antibiotics.40.612
A Herscovics. Biochim. Biophys. Acta, 1999, 1473: 96~107. doi: 10.1016/S0304-4165(99)00171-3
H Kayakiri, S Takase, T Shibata et al. J. Org. Chem., 1989, 54: 4015~4016. doi: 10.1021/jo00278a003
H Kayakiri, S Takase, T Shibata et al. Chem. Pharm. Bull. 1991, 39(6): 1378~1381. 10.1248/cpb.39.1378
(a) A D Elbein. FASEB J. 1991, 5: 3055~3063. (b) N Asano, R J Nash, R J Molyneux et al. Tetrahedron Asymmetry, 2000, 11: 1645~1680.
(a) A D Elbein, R Molyneux. J. Compre. Nat. Prod. Ⅱ, 2010, 6: 225~249; (b) P Compain, O R Martin. John Wiley & Sons Ltd: Chichester, England, 2007. 8.
(a) H Kayakiri, C Kasahara, T Oku et al. Tetrahed. Lett., 1990, 31(2): 225~226; (b) H Kayakiri, C Kasahara, K Nakamura et al. Chem. Pharm. Bull. 1991, 39(6): 1392~1396. doi: 10.1016/S0040-4039(00)94377-6
K W Hering, K Karaveg, K W Moremen. J. Org. Chem., 2005, 70: 9892~9904. doi: 10.1021/jo0516382

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
