School of Sciences, Xi'an Technological University, Xi'an 710021, China
b.
Shaanxi Key Laboratory of Optoelectronic Functional Materials and Devices, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China
c.
Institute of Environmental and Energy Catalysis, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China
d.
College of Resources and Environmental Engineering, Tianshui Normal University, Tianshui 741001, China
Received Date:
05 February 2022 Accepted Date:
23 February 2022 Revised Date:
19 February 2022 Available Online:
15 February 2023
Abstract:
In this paper, Fe36Co44 nanocluster structure is used to catalyze the hydrolysis reaction of ammonia borane to produce H2. Firstly, we complete the construction of Fe36Co44 cluster structure and calculate the electronic properties of the cluster. By comparing the adsorption process of Ammonia Borane (AB) in active sites of the cluster, which have different Effective Coordination Number (ECN), the qualitative relationship between ECN and the catalytic activation of AB is clarified, and the optimal catalytic active site is obtained. Then, from the perspective of different reaction paths, we study the hydrolysis reaction of AB in multiple paths, and obtain 5 different reaction paths and energy profiles. The calculation results show that in the case of NH bond priority break (path 5), the reaction has the minimum rate-determining step (RDS) barrier (about 1.02 eV) and the entire reaction is exothermic (about 0.40 eV). So, path 5 is an optimal catalytic reaction path. This study will have an important guiding significance for the study of the AB hydrolysis reaction mechanism.
As we all know, hydrogen (H2) [1-4] is general accepted as the clean fuel and regarded as an excellent substitute for oil fuel. However, H2 is difficult to store in compressed form or liquefied form, and this is one of the major challenges in establishing a hydrogen economy [5-8]. Ammonia-Borane (NH3BH3, AB), which has the most hydrogen-containing borohydride (19.6 wt%), high thermal stability, long-term stability in aqueous solutions and low toxicity, is becoming the leading contender for chemical hydrogen-storage materials.
Up to now, noble metal (Pt, Ru, Rh) and noble metal alloy [9-14] usually as the potential catalyst for the dehydrogenation reaction of the AB. Generally, 1 mol AB can release 3 mol H2 at ambient temperatures using an appropriate catalyst through solvolysis with protic solvents such as water and methanol. RuNi, RuPt and RuPd nanoparticles [9-11] have been proven to have high catalytic activity. Yang et al. [12]. constructed a Co1-x@Ptx core-shell structure with different ratio with carbon as the substrate, where x = 0.33, 0.43, 0.60, 0.68, 0.82. Their results show that when x = 0.68, Co0.32@Pt0.68/C structure has a higher catalytic reaction rate, reaching 4874 mL H2 min−1 gcatalyst−1 at 25 ± 0.5 ℃, which is much higher than that of pure Pt nanostructures with carbon as the substrate (2781 mL H2 min−1 gcatalyst−1). Unlike Co0.32@Pt0.68, Ru@Co [13] core–shell nanoparticles with Co atoms exposed on the outer layer can also efficiently catalyze NH3BH3 hydrolysis and more active sites deriving from the higher amorphous Co contents. CoRh nanoparticles [14] supported on the zeolitic imidazolate framework (ZIF-67) also have higher active sites. After cheap metal Co was brought into the Rh@ZIF-67, no matter how much the Co was brought, the catalytic property was increased significantly. This shows that not only precious metals (such as Pt, Ru, Rh) have higher catalytic activity, Co atoms can also efficiently catalyze AB hydrolysis. Alloys exhibit better catalytic activities than single metal for hydrogen generation from the hydrolysis of AB, which is attributed to the synergy effect between two kinds of metals.
Due to the small reserves of noble metals and high prices, the main purpose in the design of catalysts is to reduce or even eliminate noble metal materials, and use transition metal. Therefore, Co-based transition metal catalyst materials have gradually become a research hotspot, and the same is true for the process of preparing H2 by the hydrolysis of AB. It has been reported that some Co-based transition metal catalyst materials show good catalytic activity, such as CoW, CoFe, CoNi, CoCu and CoZn [15-19]. Among them, FeCo binary nanocluster structure showed the best catalytic activity and it reveals that the FeCo nanocluster structure can be used as a potential catalyst for AB hydrolysis reaction.
So far, different mechanisms have been applied to explain the hydrolysis of AB. For the Pt/γ-Al2O3 structure, it has been shown that H2O will participate in the dehydrogenation process of AB and will affect the break of the B—N bond [20]. From an experimental point of view, Chen [21] used Pt/CNT as a catalyst material, and proved that the rate-determining step (RDS), which is the step with the highest energy barrier in the reaction, is the O—H bond cleavage of water. Followed the same idea, Li [22] studied the formation process of the first H2 molecule of Pt supported on the Ni surface catalyst by the First-principle method. Their calculations show that the activation barrier is 0.75 eV and which comes from the breaking of the O—H bond in H2O. For the catalytic reaction path where Pt atoms are loaded on the surface of oxidized graphene, Wu [23] analyzed different possible reaction mechanisms, showing that the formation of the first H2 molecule is derived from the break of two B—H bond and the RDS barrier is 11.3 kcal/mol (0.49 eV).
For non-noble metal catalyst materials, Peng [24] calculations show that on the Ni2P surface, under the coordination of H2O, the breaking of the B—N bond is the RDS (0.12 eV), and the subsequent reactions are self-powered. For the Ni-Co-P surface [25, 26], the B—N bond is broken under the influence of OH− to generate *BH3OH + NH3, which becomes the RDS (1.08 eV). However, there is still a lack of research on the effect of OH− on the subsequent H2 generation process. Currently, the studies have mostly studied the catalytic process of the surface structure, and the research on the cluster nanostructures are still lacked, especially the transition metal nanocluster materials. From the perspective of computational simulation, the detailed description of the reaction process of the hydrolysis reaction, such as the interaction mechanism of the alloy components, the mode of dehydrogenation and the role of OH−, which is significant for the further design and optimization of the catalyst, is insufficient.
The Arrhenius plots can well reflect the relationship between reaction temperatures, reaction rate and activation barrier in AB hydrolysis process, and is the main method for experimental research [27-35]. There have been many correlative experiments results on binary metal alloy nanoclusters. In particular, studies on FeCo nanocluster alloy catalysts with different atomic proportions have been proposed in the literature [16]. The activation energy can be used to compare with the computational simulation.
With Co-based core-shell structure Fe22@Co58 as catalyst material [36], through the combined interaction of Co, Fe atoms, we studied the catalysis process of AB hydrolysis and the B—N, B—H and N—H bonds of AB are activated, a complete AB hydrolysis process are described. At the same time, through the introduction of OH− in the solution, greatly reduce the reaction barrier of RDS and provides an important reference for the Co-based non-noble metal catalysts designs.
In this paper, five reaction paths are summarized according to the breaking order of B—N bond, B—H bond and N—H bond in AB and the role which H2O molecule played in different reaction cases. These five reaction paths have different RDS and activation barriers. Also, the relationship between the electronic properties of the catalyst materials and the hydrolysis reaction paths has been studied. Among them, the interaction between Fe and Co atoms and the influence of OH− play a significant role, making it easier for the isolated H atoms to form H2.
All the calculations are performed by the first-principles calculation adopting density-functional theory (DFT) [37-39] using the VASP package [40, 41]. The generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) [42, 43] function is chosen to describe the exchange and correlation function. And interaction potentials of the core electrons are replaced by the Projector Augmented Wave (PAW) [44]. A plane wave basis set with a cut-off energy 400 eV is used and spin-polarization is taken into account. An ionic and electronic relaxation converged within the error of 1 × 10−3 eV/atom and the convergence precision is set to force less than 0.01 eV/Å. Also we set Smearing = 1, sigma = 0.05. Only the gamma point was used to sample the Brillouin zone of each nanoparticle and the adsorbed molecule structure [45, 46]. The Van der Waals interaction is considered in the calculation process by using DFT+D3 [47-51]. To clarify the possible reaction mechanism for Fe36Co44 nano-structure catalysis in AB hydrolysis, the energy barriers have been calculated with the climbing image nudged elastic band (CI-NEB) method [52-56]. All intermediates have no virtual frequency, and the transition state has only one virtual frequency. Device Studio [57] program provides several functions for performing visualization, modeling, and simulation.
As we all known that FeCo nanoclusters possess the high structure stability and catalytic activity at the ratio of 1:1 [19], the similar proportion of nano-cluster structure Fe36Co44 was built for studying the dehydrogenation of AB. The optimized structure of Fe36Co44 was depicted in Fig. S1 (Supporting informatin). In order to analyze the structural stability, the average binding energy (Ebind) of Fe36Co44 alloy cluster was calculated by the under formula (Eq. 1):
(1)
where Ecluster, EFe and ECo are the total energies of Fe36Co44 cluster, Fe and Co atoms, respectively. NFe and NCo are indicated as the numbers of Fe and Co atoms involved in the cluster (NFe + NCo = 80). The binding energy of Fe36Co44 nanostructure is −3.50 eV, which is leading to a stronger interatomic bond.
As shown in Fig. S1, two crystal faces (111) and (100) were exposed to the outer layer, which is known as catalytic activity surface. Generally, the different crystal single surface displayed various atomic and electric structure, which makes a difference in the molecular adsorption and the process of the catalytic transformation. It should be noted that the different crystal single surface have different kinds of atoms with different atom coordination number (CN). It should be highlighted that CN affects the adsorption and catalytic activity of catalyst. In order to further investigate the stability of the FeCo-alloy structure and its catalytic activity, the effective coordination number (ECN) [30] were calculated and expressed as Ze, n as follows ((2), (3)). And the calculation results were listed in Table S1 (Supporting informatin).
(2)
(3)
where ZS and ZI indicate the CN of the surface and inner atoms of Fe36Co44 nano-structure, respectively. We set ZI = Zb, where Zb is the CN of the bulk atoms. λ = 0 for nanoparticles. Represents the dimensions of the structure. d (=2r) is the atomic diameter of the atoms building the nanocrystal. D is the diameter of spherical nanoparticles, nanowires and the thickness of nanofilms. S is the active surface area of the cluster. PS is the packing fraction of the surface crystalline plane (the ratio of the area of the surface plane occupied by atoms to the total area of the plane). PL is the lattice packing fraction (the ratio of the volume in the unit cell of lattice occupied by atoms to the total volume of the unit cell).
As we all known, the single atom catalysis, the small cluster and the crystal single surface have the sensational catalytic activity than bulk materials, dueing to that the smaller CN leading to much dangling band. As shown in Table S1, it should be highlighted that the ECN of Fe and Co atoms in the Fe36Co44 nanostructure model are both smaller than that of the Fe and Co atoms in the bulk material. Therefore, the Fe and Co atoms with the smaller ECN in the alloy nanostructures have turned into the well catalytic active sites, especially for that of Coi atom in the tip of the Fe36Co44. As shown in Fig. S1, the surface of the cluster structure is wrapped by (111) and (100) planes and the low-index (111) and (100) planes have good stability and catalytic activity [58-64]. Due to the symmetry of the cluster structure, we only choose different adsorption positions on the (111) and (100) planes to investigate the effect of the cluster structure on AB. The related adsorption structure is shown in Fig. S2 (Supporting informatin).
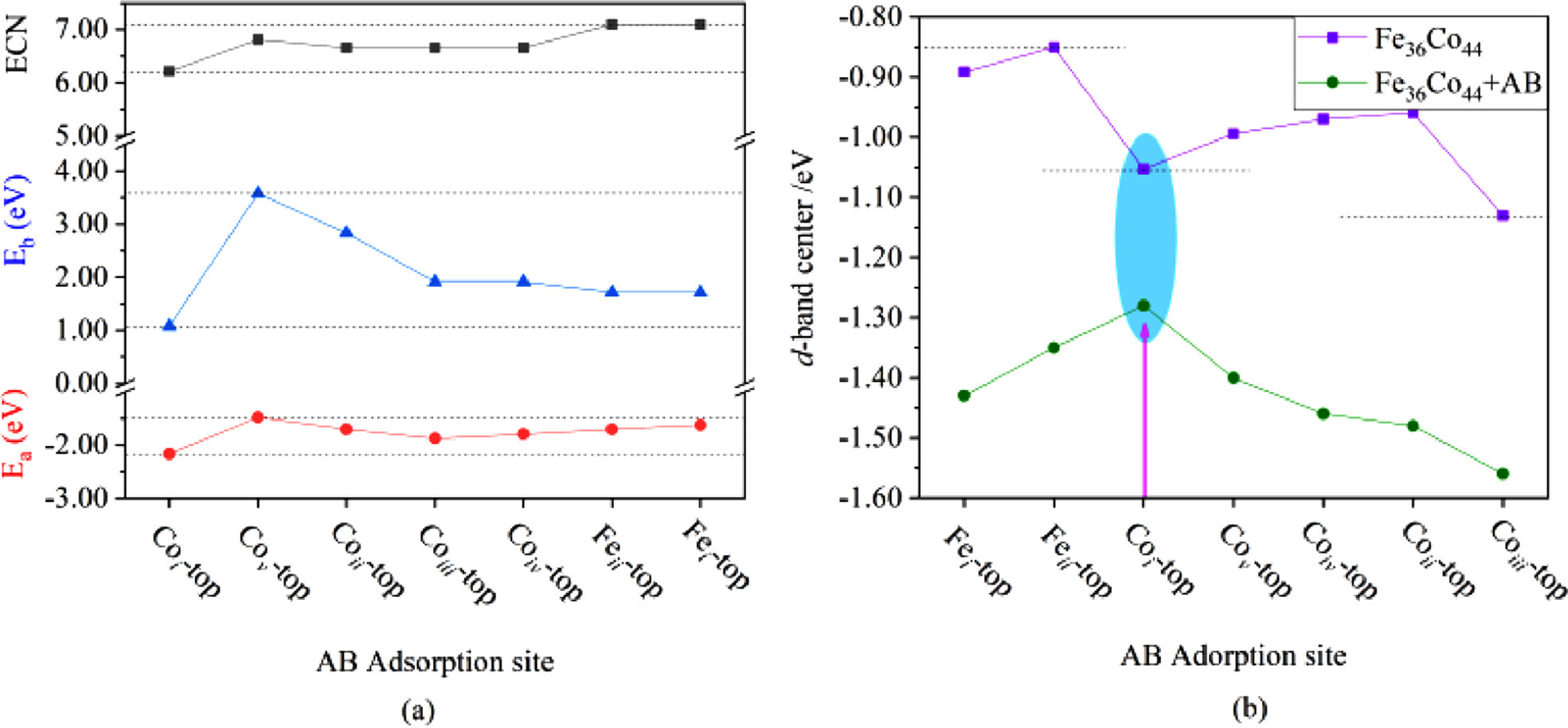
As shown in Fig. 1a, the ECN of the several adsorption sites, the corresponding adsorption energy, as well as the barrier of first dehydrogenation step was depicted clearly. It can be found that for different AB adsorption sites, when the ECN is the smallest, the breaking barrier of the B—H bond becomes the smallest, and at this time, AB has moderate adsorption energy (Fig. 1a). It shows that the adsorption of AB on Fe36Co44 cluster will affect the activity of AB hydrolysis. The smallest adsorption energy is beneficial for the hydrolysis reaction of AB.
Figure 1
Figure 1.
(a) ECN, reaction barrier energy (Eb) and AB adsorption energy (Ea). Eb is the reaction barrier to break the B—H bond (the reaction process in different AB adsorption sites, Fig. S2). (b) d-band center of different AB adsorption sites.
Generally, the location of the d-band-center is one of the critical descriptor in the process of adsorption and activation process. It can be seen from Fig. S1 that Coii and Coiv have the same physical environment and the same ECN, while Fig. 1a shows that they have different Eb, which is caused by the factors of the d-band-center in Fig. 1b. Compared with Coii, the d-band-center of Coiv is farther away from the Fermi level, indicating that this adsorption site has better oxidation. The breaking of the B—H bond requires an oxidation center. Therefore, Coiv has a lower Eb. At the same time in Fig. 1b, the adsorption site of Fe36Co44 nano-cluster Coi has a moderate d-band-center, and after adsorption of AB, the d-band-center increases and becomes closest to the Fermi level (Table S2 in Supporting informatin for specific values). The rise of d-band-center relative to the Fermi level will lead to a mildly rise of the antibonding states between the B/N 2p states and the metal d states, generating a moderately stronger metal-B/N bond. This indicates that the Coi adsorption site have stronger interaction and can better activate AB. The electron density at the Fermi level decreases with decreasing d-band-center, and the electron density at the Fermi level is closely related to the progress of the chemical reaction. Therefore, we selected the Coi-top position as the initial structure for the subsequent AB hydrolysis to produce H.
Firstly, AB dehydrogenation and H2O dissociation processes are successively performed on the surface of the Fe36Co44 cluster (Fig S2). The H2O molecule, the one that dissociated, did not interact with AB.
As shown in Table S3 (Supporting informatin), the B—H bond becomes longer due to the interaction of adsorbed AB and the Fe36Co44 cluster, which indicates that maybe it is easily to be broken. As a possibility, we designed the reaction process as shown in Fig. S3 (Supporting informatin). H2O molecule (I → II) and AB molecule (II → III) dissociate on the surface of Fe36Co44 cluster respectively. The dissociation barrier of H2O molecule is 1.20 eV and the reaction heat is 1.10 eV, according to a very rough rule-of-thumb indicating that this reaction step can be carried out at about 480 K [65-68]. Then, the B—H bond break process in AB was carried out, and the reaction barrier was still large at 1.65 eV. The energy released from structure TS-1 to structure II (1.10 eV) is not sufficient to overcome the reaction barrier from structure II to structure TS-2, indicating that this is a reaction step that cannot proceed spontaneously. The B—H bond breaking process as the RDS of the reaction process prevents the reaction from continuing. This indicates that H2O molecules can spontaneously dissociation under moderate reaction conditions, while B—H bond break is affected by external conditions, such as H2O molecule. Considering this effect, we built an AB + H2O co-adsorption structure to break the B—H bond or N—H bond and achieve AB hydrolysis generates H.
We investigated the co-adsorption of AB and H2O on the surface of Fe36Co44, and the calculation results are listed in Table S3. After AB + H2O is co-adsorbed on the surface of Fe36Co44, the B—H bond becomes longer (from 1.208 Å to 1.386 Å), which indicates that it is easily activated.
As shown in Fig. S4 (Supporting information), under the premise of the interaction between H2O and AB, the break of the B—H bond becomes easy, and the reaction barrier and reaction endotherm are 0.80 and 0.55 eV respectively, which is much smaller than that of the B—H bond break in Fig. S3. Therefore, the direct interaction between H2O molecules and AB is very significant during the AB hydrolysis. In the next process of breaking the N—H bond, the reaction barrier is about 1.04 eV and the reaction endotherm is 0.25 eV, which indicates that it is more difficult to break the N—H bond than the B—H bond at this time. From structure III, after two isolated H atoms obtained, to the structure V that finally forms H2 molecules, absorbed a lot of heat, indicating that the reaction was difficult to proceed. The main reason is that the adsorption energy of H atoms on Fe atoms is too large, which obstruct the formation of H2 molecules.
Similar results showed that the N—H bond may be broken firstly (the bond length becomes longer after adsorption). As shown in Fig. S6 (Supporting information), H2 produced by the process of hydrolysis reaction of AB. The reaction process is N—H bond and B—H bond broken, and then isolated H atoms form H2. Firstly, the barrier to break the N—H bond is about 1.20 eV, while the barrier to break the B—H bond is about 0.80 eV. During the subsequent process of breaking the B—H bond, the reaction barrier is about 0.68 eV, this is close to the barrier of B—H bond break. It shows that it is easier to break the B—H bond firstly rather than to break the N—H bond firstly and no matter whether the B—H bond or the N—H bond is broken firstly, the reaction barrier of breaking the B—H bond or the N—H bond is basically the same. That is to say, the breaking order of the B—H bond and N—H bond basically does not affect the value of their breaking barrier. Observing that Figs. S5 and S6 (Supporting information), it can be found that even though the formation of H2 is a large endothermic step, the whole reaction can still proceed spontaneously at room temperature.
From the perspective of bond energy, we can find that the bond energy of B—N bond is weak, and it is also easy to break firstly in the catalytic process. Therefore, we calculated the reaction path where the B—N bond was firstly broken under H2O co-adsorption. This process only needs to overcome a small energy barrier (0.47 eV), as shown in Fig. S5. The entire reaction process of one H2 molecule produced is described in Fig. S5. After breaking the B—N bond, the next key step is the dissociation of H2O molecules. From structure III to structure IV, the cleavage of O—H bond in H2O molecules on the surface of the Fe36Co44 cluster was completed, and the cleavage barrier was about 0.90 eV, which provided one H atom for the later formation of H2. During the reaction, the B—H bond breaking process with the largest potential barrier, was about 1.70 eV, which was RDS and shows that the process is difficult to carry out. Therefore, the case where the B—H bond breaks before the N—H bond is disadvantageous.
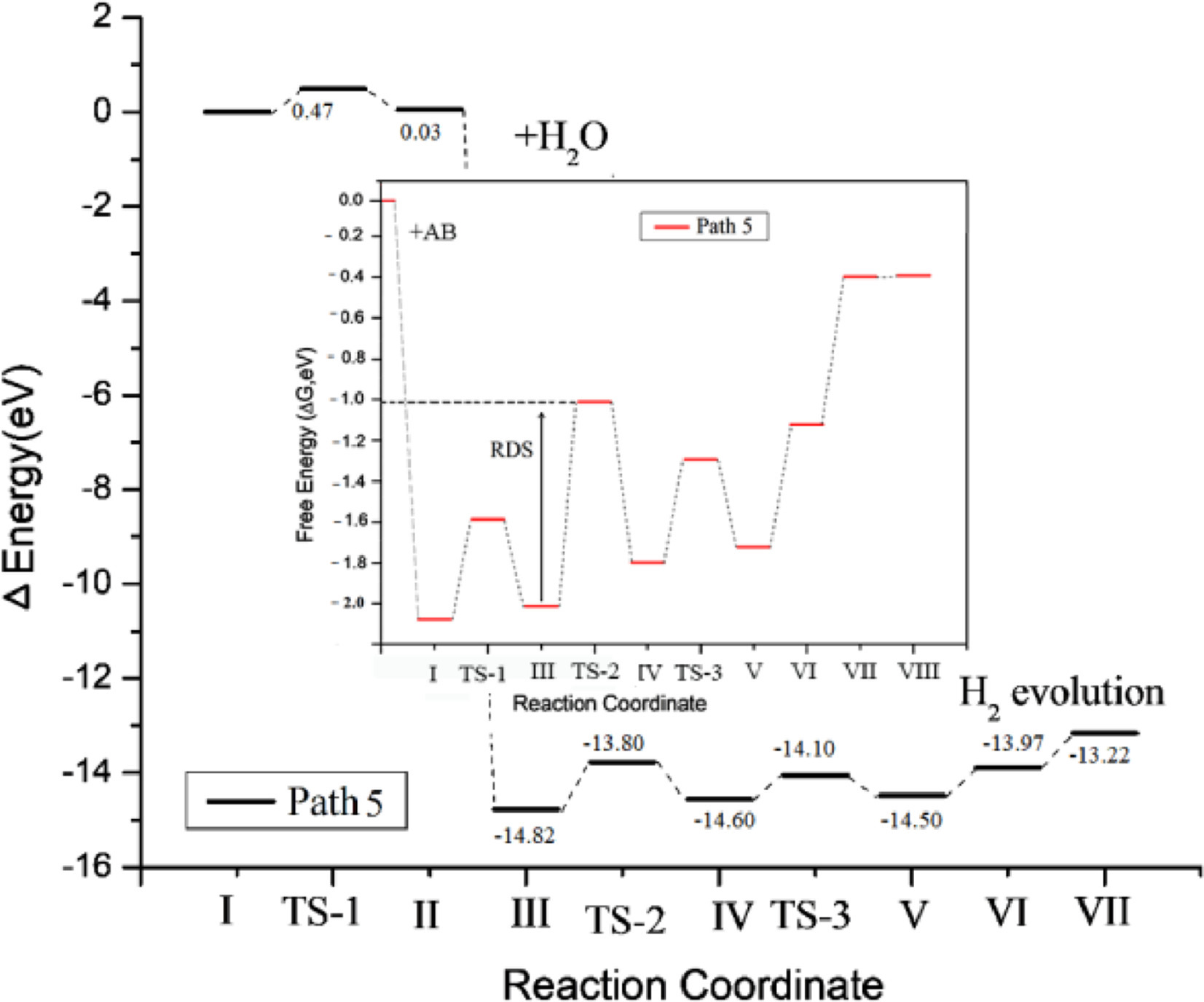
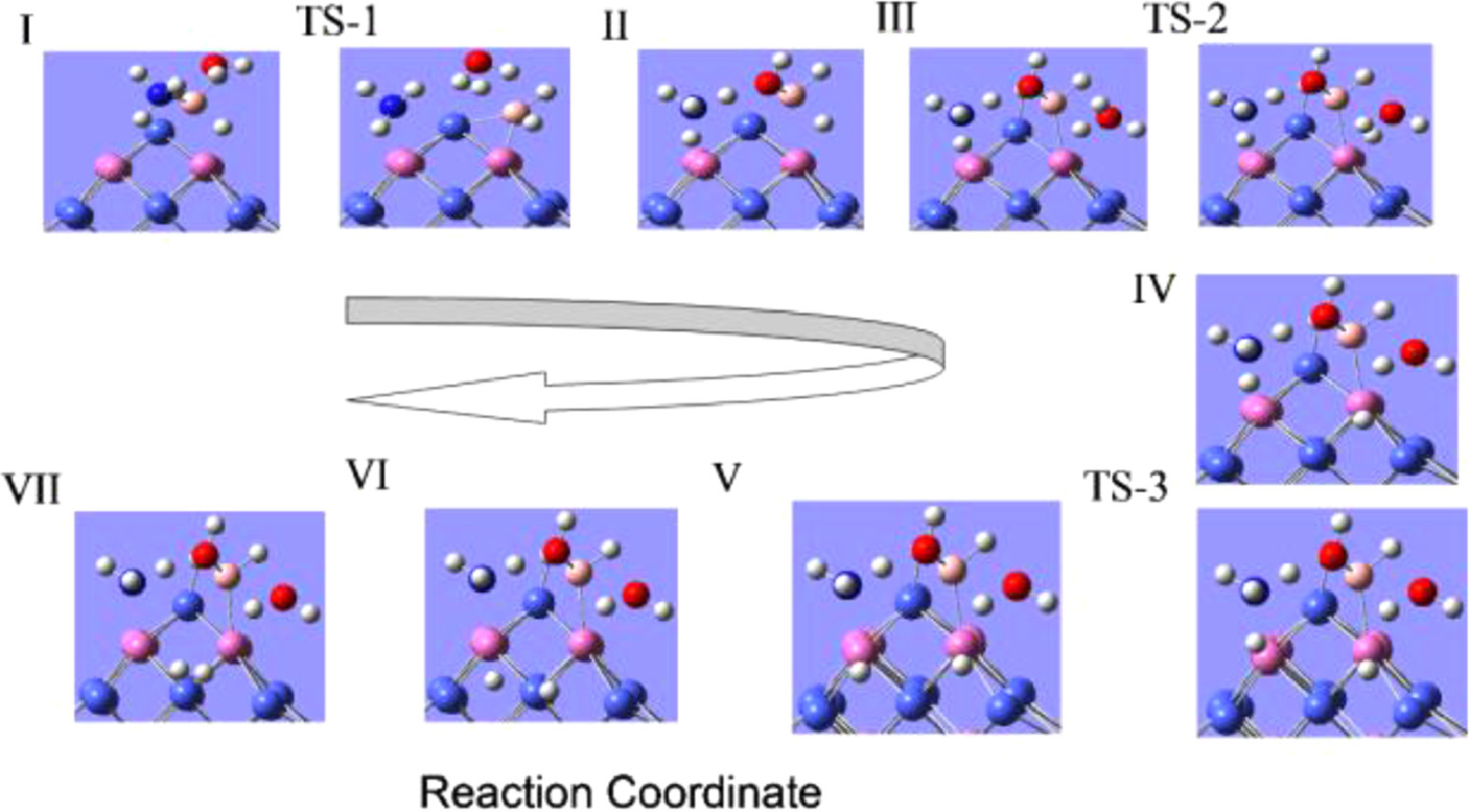
As shown in Fig. S5, during the reaction, the breaking barrier of the B—H bond is large, so we try to break the N—H bond to provide the H atom after the H2O molecule is dissociated (from structure IV to V). It can be found that adopt the reaction path shown in Fig. 2, the potential barrier of the entire reaction process is relatively low, that is, the RDS of the reaction is lower. The results of free energy change (ΔG) of the whole reaction process have shown that the potential barrier of this step, break the N—H bond, is 0.52 eV, which is greatly reduced compared with the potential barrier required to break B—H bond. Under these conditions, the dissociation process of H2O (III→IV) molecules has become the RDS of the whole reaction, and the barrier is about 1.02 eV. It is proved that on the surface of FeCo alloy clusters, the main limiting factor of the hydrolysis reaction of ammonia borane is the dissociation of H2O. The whole reaction is exothermal and the RDS is 1.02 eV, which is a small value, and so, the reaction is easier to carry out, even at room temperature. The step by step process of the reaction is represented in Scheme 1. So, when B—H bond break, we believe that path 5 is the more possible reaction path. This conclusion is consistent with the existing literature, and is beneficial to the design of FeCo catalysts with different morphologies and different atomic ratios.
Figure 2
Figure 2.
AB hydrolysis reaction path 5 and calculation free energy (ΔG) profiles.
Throughout the description of the reaction process of the five hydrolysis mechanisms, we found that the RDS of path 3 and path 5 is about 1.0 eV. This shows that for the entire reaction process, when the ambient temperature is slightly higher than room temperature, both path 3 and path 5 could happen spontaneously. Between 293~313 K, experimental studies have shown that as the reaction temperature increases, the rate of H production gradually increases and the activation energy is gradually decreasing.
The coordination effect between the two elements of the alloy nanostructure plays a key role in promoting the performance of the catalyst metal atom Fe, Co and Cu have different effects on AB adsorption and activation, and this effect can be coordinated. Similarly, in this paper, the interaction between Fe and Co atoms plays an important role in the dehydrogenation and the formation of H2.
The structure of the surface d-band-center is a critical descriptor in the adsorption properties of many simple adsorbates. In Table S1, we listed the d-band-center of Fe and Co atoms in bulk materials and alloy nanostructures. The adsorption structure is shown in Fig. S8 (Supporting information). Compared with bulk materials, alloy nanostructures have higher d-band-center, which indicates that the electron locality of Co/Fe atoms is stronger. After AB is adsorbed on Fe36Co44 nano-structure, it is proposed that the mildly rise of d-band center relative to the Fermi level will lead to a mildly rise of the antibonding states between the B/N 2p states and the metal d states, generating a moderately stronger metal-B/N bond. It strengthens the adsorption of AB, promotes the transfer and exchange of electrons, and facilitates the decomposition of AB. The electron density at the Fermi level decreases with decreasing d-band-center, and the electron density at the Fermi level is closely related to the progress of the chemical reaction.
At the Coi-top adsorption site, the bond energy changes of AB are shown in Fig. S7 (Supporting information). By observing PDOS and -COHP, it can be found that after adsorbed by nanoclusters, the orbital hybridization between B and H atoms becomes more weakly. At the same time, the anti-bonding orbital of the B—H bond is also occupied by electrons. Otherwise, the anti-bonding orbital of the N—H bond does not appear to be occupied by electrons, indicating that the N—H bond is not significantly weakened and the B—H bond is greatly weakened during the adsorption process. In Fig. S7a, the anti-bonding orbit of the B—N bond is occupied by some electrons and arise a small peak, which weakened the bond energy of B—N. Therefore, the breaking of the B—H bond and B—N bond is a priority step.
As shown in Fig. S7, we calculated the changes of bond length and bond angle of AB after and before adsorption. It can be seen that the B—N bond changes greatly, indicating that the B—N bond has been better activated. After adsorption, B—H becomes larger, about 11.6%, while the N—H bond is only lengthened by about 1.8%. The bond angle changes have the same law, and the H—B—H bond angle changes greatly.
The integration of -COHP (-ICOHP) up to the Fermi energy quantitatively describes the bonding strength. After adsorption, the B—H bond changed from 3.84 eV to 3.54 eV, indicating that the orbitals of H and B overlapped better than after adsorption (Table S4 in Supporting information for specific value). It is proved that the adsorption process weakens the B—H bond. Combined with the changes in bond length and bond angle in Table S3, it can be explained that in the subsequent dehydrogenation process, the breaking of the B—H bond will be easier than the breaking of the N—H bond, if the B—N bond does not break.
Experimental studies show that in the presence of NaOH, the rate of AB hydrolysis reaction will be greatly accelerated [69-74]. And the AB solution is alkaline (pH ≈ 9.1), so the OH− present in the solution will affect the progress of the reaction. This effect is achieved by *OH− adsorption on the surface of Fe36Co44 catalyst. The red parts in Fig. S6 indicate the reaction process of the H2 formation process after the adsorption of 2*OH−. The potential barrier is about 0.27 eV, which greatly reduces the difficulty of H2 formation. The adsorption of 2 *OH− makes the process of forming H2 from isolated H atoms easier, because the self-charge distribution of H atoms and interaction of H-Fe (Co) have changed. And the H atom charge distribution and H-Fe (Co) interaction are described in the following.
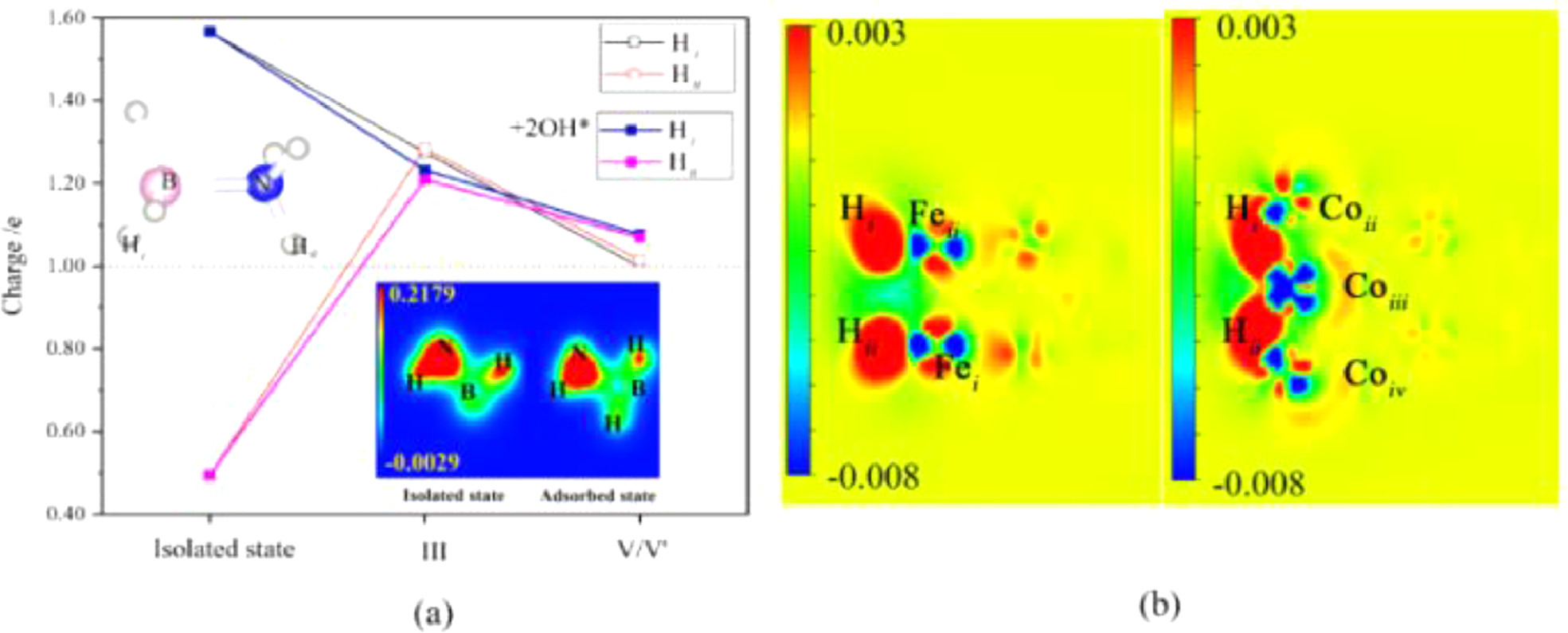
The charge densities of the isolated state and adsorbed state (Fig. 3a) also show the weakening of the B—H bond and the N—H bond is still relatively strong. Therefore, the reaction barrier to break the B—H bond is lower than the reaction barrier to break the N—H bond. Fig. 3b shows the difference charge density under the influence of 2*OH−, indicating a strong interaction between two H atoms, and at this moment Co atoms become a bridge for charge transfer.
Figure 3
Figure 3.
(a) H atom charge distribution of isolated state, III and V/V' structure and (b) V/V' structure difference charge density (the atom labels are from Fig. S1). The default isovalue is 0.02 e/Å3.
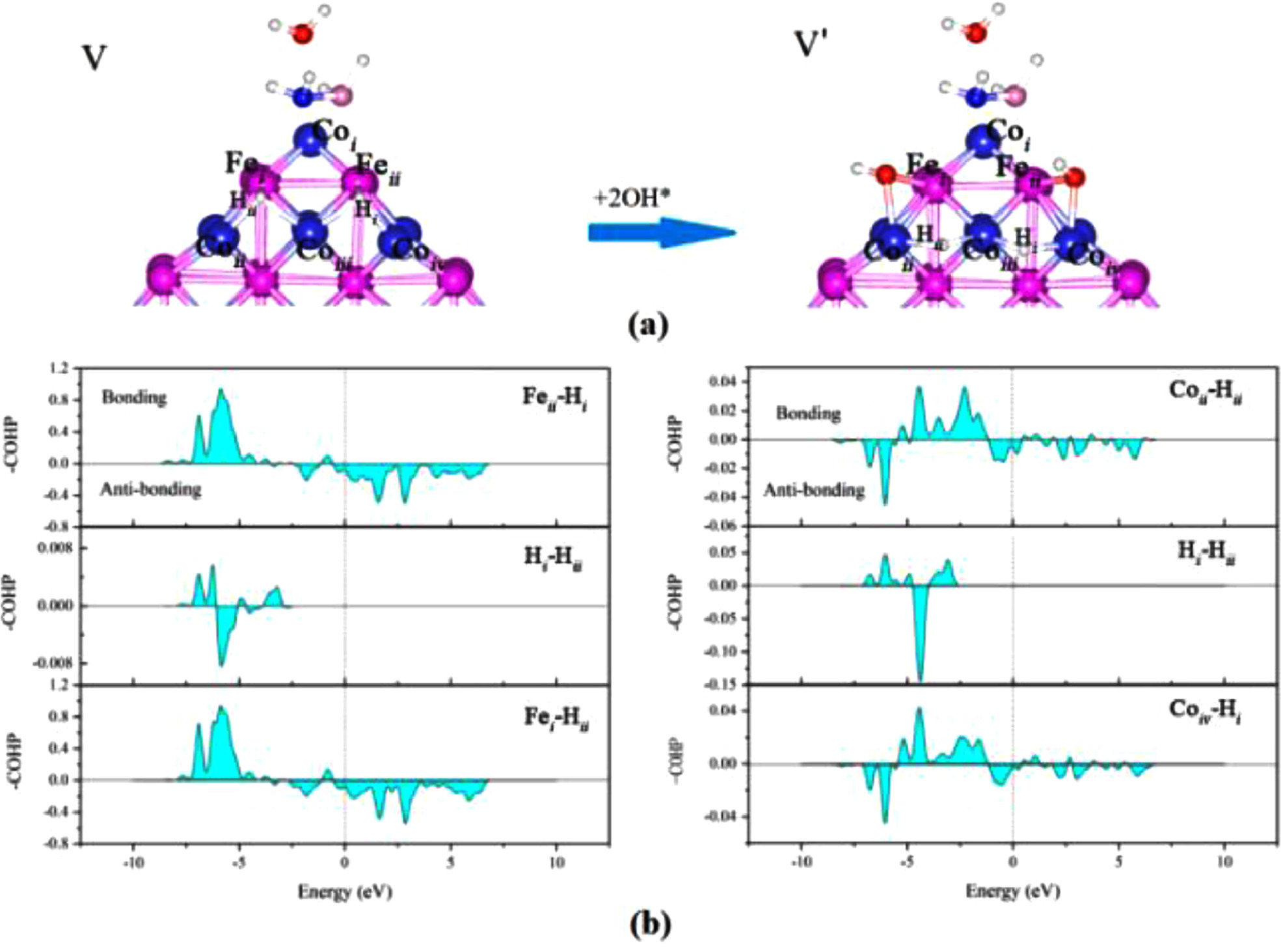
As shown in Fig. 4, under the effect of 2*OH−, the position of the isolated H atom changed, isolated H atoms are adsorbed on the bridge site of two Co atom, and the adsorption structure changes from V to V'. Comparing 2*OH− before and after adsorption, we found that the structure of Co-H bonds after adsorption is weaker than that of Fe-H bonds before adsorption. As shown in Fig. 4a, Hii-Fei bond length increase from 1.75 Å to 2.90 Å, Hi-Feii bond length increase from 1.75 Å to 3.03 Å, and Hi-Coiii bond length decrease from 1.84 Å to 1.64 Å. Otherwise, Hii-Coii and Hi-Coiv bond lengths remain the same, about 1.70 Å. This indicates that the H-Fe interaction is weakening while the H—Co interaction is strengthening. Farther more, this confirms that Coiii acts as a bridge for the exchange of charge between H atoms. The H atoms, at this time, are easily separated from the cluster surface to form H2.
Figure 4
Figure 4.
(a) *OH− adsorption effect on the structure V and (b) atom –COHP of the structure V.
From Fig. 3a, it can be seen that charge change of the H atom during the drop process: one increases electrons, one loses electrons, and at the same time undergoes oxidation and reduction reactions, and eventually they are all electrically neutral. In Fig. 3b, the charge density 2D plot indicates that Co atom builds a bridge for charge transfer of two isolated H atoms. The interaction between two isolated H atoms is strong, and it is easier to form H2.
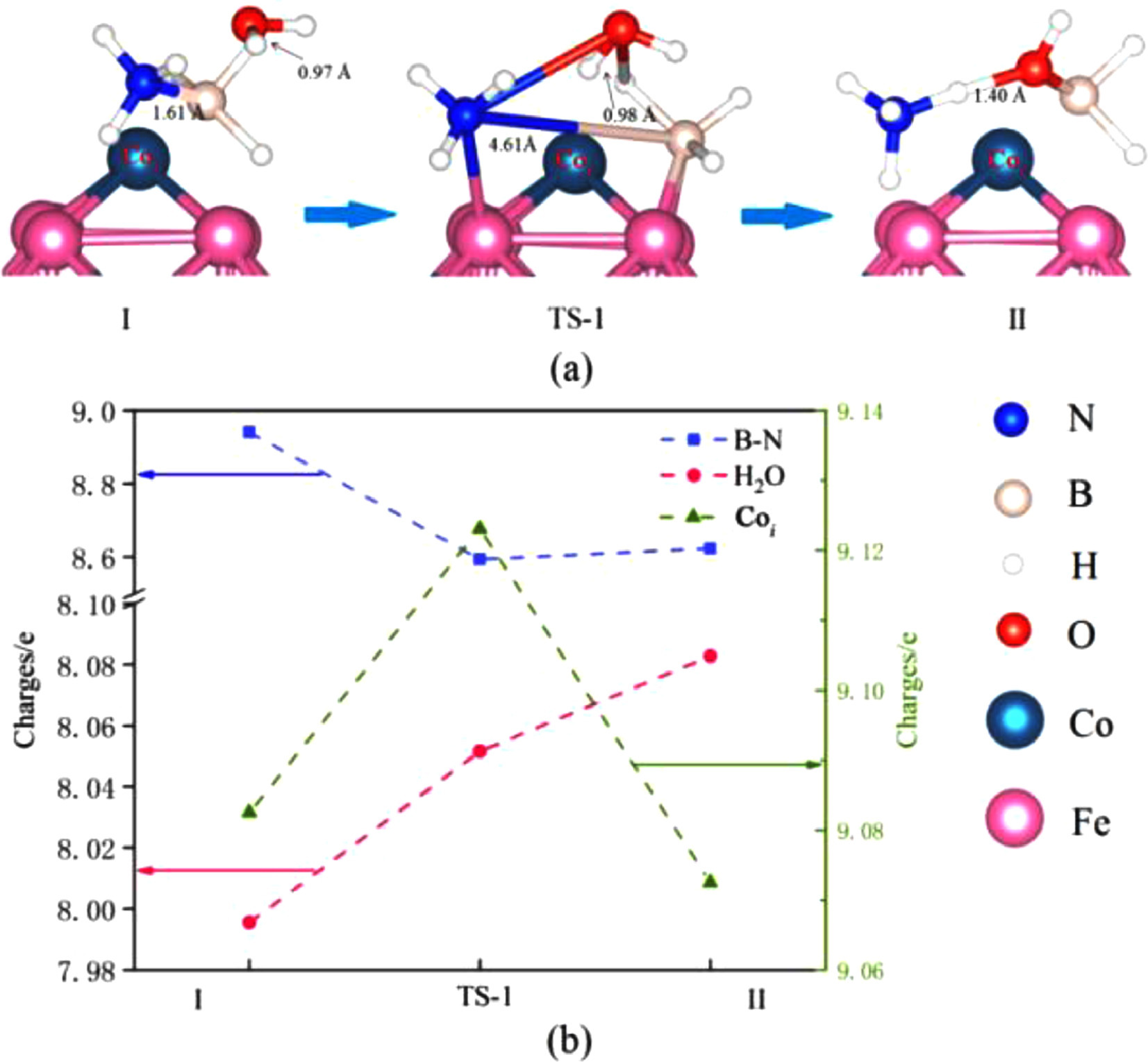
From the perspective of bond energy, we can find that the bond energy of B—N bond is weak, and it is also easy to break firstly in the catalytic process. Therefore, we calculated the reaction path where the B—N bond was firstly broken under H2O co-adsorption. This process only needs to overcome a small energy barrier, as shown in Fig. 2. After the B—N bond breaken, H2O molecule is inserted between B atom and N atom to form small molecular groups with AB, as shown in Fig. 5a structure II. At this time, the O—H bond in the H2O molecule has been broken, and the length of one O—H bonds becomes 1.40 Å. Such molecular groups are conducive to the release of H atoms. In response to this process, Fig. 5b describes the change in charge. In the process of breaking the B—N bond, B—N as a whole loses electrons and the interaction is weakened. In the transition state (TS-1) of the process, the electrons lost by B—N accumulate on the surface of Co atoms and H2O molecules, leading to the breaking of H—O bonds in H2O molecules.
Figure 5
Figure 5.
(a) B—N bond breaks process and (b) charge distribution of AB under the influence of H2O molecules.
The breakage of B—N bond provides favorable conditions for the subsequent acquisition of isolated H atoms and promotes the extension of B—H bond and N—H bond. Thus, this process becomes a prerequisite for path 4 and path 5 to occur, resulting in the reaction path with minimal activation barrier, and is a critical step in AB hydrolysis.
In this paper, we performed a theoretical calculation simulation study on the generation of H2 by Fe36Co44 alloy cluster-catalyzed hydrolysis of AB. Through the adsorption process of different active sites, the qualitative relationship between ECN and activation process of AB was obtained and we find that Coi atoms with the lowest ECN have the lowest activation barrier, is the best active site. Throughout the calculation process, we described 5 different possible reaction paths and, of course, obtained different RDSs. Among of them, we obtain the catalytic reaction path with the smallest RDS and the capable of self-powered at room temperature. For the optimal reaction path, path5, RDS is 1.02 eV. The presence of *OH−changes the adsorption energy of H atoms on the surface of the alloy clusters, making the process of H atoms forming H2 easier. According to the present of *OH− in the reaction process, the activation barrier decrease from 1.56 eV to 0.27 eV, which can significantly reduce the reaction temperature and accelerate the reaction rate. It lays a foundation for further exploring the transition metal-catalyzed AB hydrolysis.
Declaration of competing interest
The authors declare that they have no known competing financial interests.
Acknowledgments
This study was funded by the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0676) and the National Supercomputing Center in Zhengzhou. Jinrong Huo was partially supported by the postgraduate research opportunities program of HZWTECH (No. HZWTECH-PROP).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.066.
Hongzhiwei Technology, Device Studio, Version 2021A, China, Available online, 2021, https://iresearch.net.cn/cloudSoftware (accessed on 2021/2008/2001).
Figure 1
(a) ECN, reaction barrier energy (Eb) and AB adsorption energy (Ea). Eb is the reaction barrier to break the B—H bond (the reaction process in different AB adsorption sites, Fig. S2). (b) d-band center of different AB adsorption sites.
Figure 3
(a) H atom charge distribution of isolated state, III and V/V' structure and (b) V/V' structure difference charge density (the atom labels are from Fig. S1). The default isovalue is 0.02 e/Å3.

 DownLoad:
DownLoad:

 下载:
下载:







