Citation:
Wenwen Li, Ge Feng, Jia Liu, Xing Zhong, Zihao Yao, Shengwei Deng, Shibin Wang, Jianguo Wang. The Structural and Chemical Reactivity of Lattice Oxygens on β-PbO2 EOP Electrocatalysts[J]. Chinese Journal of Structural Chemistry,
2022, 41(12): 221205.
doi:
10.14102/j.cnki.0254-5861.2022-0153
The Structural and Chemical Reactivity of Lattice Oxygens on β-PbO2 EOP Electrocatalysts
作者简介:Wenwen Li is currently a PhD candidate in the College of Chemical Engineering at Zhejiang University of Technology, China. Her research interests are the processes of electrochemical ozone production based on DFT calculations; Ge Feng is currently a PhD candidate in the College of Chemical Engineering at Zhejiang University of Technology, China. Her research focuses on electrochemical water splitting reaction based on real electrocatalytic conditions; Jia Liu is a PhD candidate in the School of Chemical Engineering, Zhejiang University of Technology, China. She is passionate about electrochemical ozone generation (EOP) research under neutral conditions, with a particular focus on the development of advanced electrocatalysts for EOP and the promotion of their industrial application; Xing Zhong is a professor of College of Chemical Engineering at Zhejiang University of Technology, China. In recent years, he is mainly engaged in the research of green industrial micro-reaction engineering, and has accumulated rich experience in the development and industrialization of PEM electrolytic ozone generator; Zihao Yao is an instructor of College of Chemical Engineering at Zhejiang University of Technology, China. His research involves the quantitative determination of C-C coupling mechanisms and detailed analyses on the activity and selectivity for Fischer-Tropsch synthesis using microkinetic modeling with coverage effects; Shengwei Deng is an associate professor of College of Chemical Engineering at Zhejiang University of Technology, China. He is mainly engaged in polymer synthesis, multiscale simulation of structure and interface properties of supported catalysts based on multi-scale simulation method; Shibin Wang is a lecture of College of Chemical Engineering at Zhejiang University of Technology, China. He completed his PhD in 2018 at University of Chinese Academy of Sciences and worked as the postdoc fellowship during 2018~2020 in Tsinghua University, China. He is currently engaged in the theoretical calculation of photocatalysis and electrocatalysis; Jianguo Wang is a professor and the head of College of Chemical Engineering at Zhejiang University of Technology, China. He completed his PhD in Chemical Technology in 2004 at Tianjin University, China. Prior to that, he received his Master of Chemical Engineering from Nanjing Tech University, China. He leads a research group focusing on design, preparation and application of nano-micro catalysts based on theory and experiment. In addition to authoring 100+ publications, he collaborated with numerous companies and is committed to industrializing his research results;
Institute of Industrial Catalysis, State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310032, P.R. China
Author Bio:Wenwen Li is currently a PhD candidate in the College of Chemical Engineering at Zhejiang University of Technology, China. Her research interests are the processes of electrochemical ozone production based on DFT calculations;Ge Feng is currently a PhD candidate in the College of Chemical Engineering at Zhejiang University of Technology, China. Her research focuses on electrochemical water splitting reaction based on real electrocatalytic conditions;Jia Liu is a PhD candidate in the School of Chemical Engineering, Zhejiang University of Technology, China. She is passionate about electrochemical ozone generation (EOP) research under neutral conditions, with a particular focus on the development of advanced electrocatalysts for EOP and the promotion of their industrial application;Xing Zhong is a professor of College of Chemical Engineering at Zhejiang University of Technology, China. In recent years, he is mainly engaged in the research of green industrial micro-reaction engineering, and has accumulated rich experience in the development and industrialization of PEM electrolytic ozone generator;Zihao Yao is an instructor of College of Chemical Engineering at Zhejiang University of Technology, China. His research involves the quantitative determination of C-C coupling mechanisms and detailed analyses on the activity and selectivity for Fischer-Tropsch synthesis using microkinetic modeling with coverage effects;Shengwei Deng is an associate professor of College of Chemical Engineering at Zhejiang University of Technology, China. He is mainly engaged in polymer synthesis, multiscale simulation of structure and interface properties of supported catalysts based on multi-scale simulation method;Shibin Wang is a lecture of College of Chemical Engineering at Zhejiang University of Technology, China. He completed his PhD in 2018 at University of Chinese Academy of Sciences and worked as the postdoc fellowship during 2018~2020 in Tsinghua University, China. He is currently engaged in the theoretical calculation of photocatalysis and electrocatalysis;Jianguo Wang is a professor and the head of College of Chemical Engineering at Zhejiang University of Technology, China. He completed his PhD in Chemical Technology in 2004 at Tianjin University, China. Prior to that, he received his Master of Chemical Engineering from Nanjing Tech University, China. He leads a research group focusing on design, preparation and application of nano-micro catalysts based on theory and experiment. In addition to authoring 100+ publications, he collaborated with numerous companies and is committed to industrializing his research results;
Received Date:
13 June 2022 Accepted Date:
23 August 2022 Available Online:
02 December 2022
Abstract:
The oxygen evolution reaction (OER) and electrochemical ozone production (EOP) attracted considerable attention due to their wide applications in electrocatalysis, but the detailed reaction mechanism of product formation as well as the voltage effect on O2/O3 formation still remains unclear. In this work, density functional theory calculations were used to systematically investigate the possible reaction mechanisms of OER and EOP on the PbO2 (110) surface, with the possible reaction network involving surface lattice oxygen atoms (LOM) proposed. The results show that the LOM-2 reaction pathway involving two surface lattice oxygen atoms (Olatt) and one oxygen atom from H2O was the most thermodynamically reactive. Different potential determining step (PDS) was obtained depending on the multiple reaction pathway, and the results show that the facile diffusion of Olatt would proceed the LOM pathway and promote the formation of surface oxygen vacancies (Ovac1/Ovac2). Furthermore, Ovac1/Ovac2 formation on the surface would trigger further reactions of H2O adsorption and splitting, which refilled the oxygen vacancy and ensured the considerable stability of the PbO2 (110) surface. Multiple H2O dissociation pathways were proposed on PbO2 (110) with oxygen vacancy sites: the acid-base interaction mechanism and the vacancy fulfilling mechanism.
As one of the most environmental-friendly oxidants, ozone (O3) has considerable oxidation potential of 2.07 V[1] and wide applications in organic synthesis, [2-4] sterilization, [1] disinfection, [5] etc. The electrochemical ozone production (EOP) route was considered to be the promising method for O3 production, in which process H2O molecules split and were oxidized on the anode to form the O3 product. The EOP attracted considerable attention due to its advantages including simple equipment requirement and harmful byproducts reduction.[6] However, the EOP efficiency was usually limited due to the competitive reaction pathway of oxygen evolution reaction (OER).[6-11] The OER process was generally considered to be more favorable than the EOP because of the lower anodic potential of O2 formation. To promote the selectivity of O3 under electrolysis, effective strategies are required to substantially elevate the OER over-potential, thus reducing the competitive O2 generation. In-depth understanding for the reaction mechanisms of O2/O3 formation is therefore required to reveal the key factors in affecting the over-potential and provide insights in regulating the O2/O3 selectivity.
With an intention to promote the inherent molecular oxygen adsorption, various catalysts including Pt, [12] PbO2, [13] SnO2, [14,15] and boron-doped diamond (BDD) were used for the feasible EOP.[16,17] Within the above mentioned catalysts, PbO2 was most widely used as the electrode material in electrochemical reactions including OER[18-20] and EOP[11,13] due to its good conductivity, stability, and low cost.[21] Previous work by Zhong and coworkers also reported a novel β-PbO2 nanorod (NR) exposing (101) and (110) facet as the effective EOP electrocatalyst.[22]
The adsorbate evolution mechanism (AEM) was mostly adopted for the OER and EOP mechanisms, which follows the four- and six-proton couple electron-transfer (PCET) mechanisms in OER and EOP processes, respectively. In AEM mechanism, the adsorbed reaction intermediates including OH*, OOH*, O*, O2*, O2*+OH*, O2*+O* and O3* are suggested to originate from H2O.[23-29] In addition to the AEM mechanism, the lattice oxygen oxidation-mediated mechanism (LOM) was proposed in recent years, whereby the O2 product was considered to originate from the surface lattice oxygen.[22,30-32] Mefford and coworkers have proved the participation of lattice oxygen by ab initio modeling in the Sr2+ doped cobaltite perovskites La1-xSrxCoO3-δ catalyst, in which the Co-O covalent interaction as well as surface oxygen vacancy concentration was modified by Sr2+ doping.[31] Zhong et al. reported the spontaneous LOM reaction pathway on the β-PbO2 nanorod electrocatalyst exposing (101) and (110) surface.[22] In situ 18O isotope-labeling differential electrochemical mass spectrometry (DEMS) experiments in combination with DFT revealed the possibility of LOM reaction mechanism, with a moderate energy barrier for lattice oxygen diffusion predicted on the (101) and (110) surfaces (0.74 and 1.25 eV). Wang and coworkers further investigated the AEM/LOM mechanism thoroughly on the PbO2 (101) surface, showing that LOM reaction pathway is more favorable than the AEM reactions mainly attributed to the weak Pb-O covalent interactions and facile lattice oxygen diffusion. Theoretical overpotential within the LOM mechanism was calculated to be 0.48 V for the O3 formation. Although the detailed reaction mechanism has been fully investigated on the PbO2 (101) surface, the understanding of O2/O3 selectivity with the effect of multiple reaction mechanisms still remains insufficient.[33]
In this work, the PbO2 (110) surface was used as the catalyst model for theoretical investigations of the OER/EOP mechanism. DFT calculations were performed to give comprehensive understandings of the reaction mechanism. DFT in combination with the ab initio molecular dynamic (AIMD) simulations revealed that the O3 formation is dominated by the LOM-2 mechanism, in which two surface lattice oxygens and one oxygen from H2O participated in the formation of O3. The PbO2 (110) surface lattice oxygen is predicted to be readily migrated and desorbed from the surface to form oxygen vacancies due to the weak Pb-O covalent interaction. H2O adsorption and splitting subsequently occurred on the PbO2 (110) surface with oxygen vacancies, resulting in the fulfilling of oxygen vacancies. The reaction network involving surface lattice oxygen consuming and refilling via the OER/EOP and H2O dissociation was proposed.
COMPUTATIONAL DETAILS
Periodic DFT calculations were performed by Vienna ab initio simulation package (VASP), [34,35] with exchange correlation function of Perdew-Burke-Ernzerhof (PBE)[36] and the Projected Augmented Wave (PAW) method[37,38] applied to describe the ion-electron interaction. The cutoff energy for the plane wave basis sets was 400 eV. The convergence criteria of geometry and energy were set to be 0.02 eV/Å and 10-6 eV, respectively. For O and Pb elements in the PbO2 (110) surface, O2s2p and Pb6s6p5d were treated as the valence state, respectively. The climbing image nudged elastic band (CI-NEB) approach[39,40] in combination with the dimer method[41-44] was applied to search the transition states. Harmonic frequencies of the adsorbates in the local minima and transition states were calculated with a much tighter energy convergence of 10-6 eV, and zero-point energy (ZPE) corrections were included in the calculated energy barriers and reaction energies.
The PbO2 (110) surface was constructed upon the optimized β-PbO2 with space group P42/mnm, in which the lattice constant of the bulk phase was calculated to be a = b = 5.08 Å, c = 3.45 Å.[33] The symmetric and stoichiometric PbO2 (110) surface was constructed with the supercell of p-(3 × 2) and the thickness of four Pb12O24 atomic layers, and the lattice constant value for the slab model was predicted to be 10.37Å × 14.31Å × 28.48Å. The Brillouin zone of PbO2 was sampled by Monkhorst-Pack k-points grids of (2 × 2 ×1). The vacuum space is larger than 15 Å to avoid the interactions between adjacent PbO2 (110) slabs. Owing to the considerable computational cost resulted from the large cell, the bottom half layers of the slab model were kept constrained in their bulk phase throughout the calculations.
The Gibbs free energies for each elementary step were calculated by: G = Eelect + EZPE - TS, where Eelect is the electronic energy (0 K) by DFT calculations, and EZPE is the zero-point energy. For surface-adsorbate species, all 3N degrees of freedom of the adsorbates were treated as harmonic vibrations, and the entropy was calculated as a sum of the contributions from these vibrational motions. In the initial stage of H2O adsorption and splitting, the gaseous H2O molecule was adsorbed on the surface, and the entropy of H2O molecule was treated by the standard entropy (S (T, P°)), which was calculated by Shomate equation in the NIST-JANAF thermochemical table. Under basic conditions (pH > 7), hydroxide anions are oxidized and two/three H2O molecules are produced along with O2/O3. To make a connection to experiment, in which the standard hydrogen electrode was applied as the reference, the adsorption energies of intermediates are referenced to the reservoir species of H2O and H2 in the model by Man et al.[25] Free energies for hydroxide ion (OH-) were treated as GH2O - 1/2GH2. Adsorption energies for reaction intermediates are defined as follows:
where G*, GOH*, GO*, GOOH*, G(O2* + OH*) and G (O2* + O*) are the free energies of *, OH*, O*, OOH*, O2*+OH* and O2*+O*, respectively; GH2O and GH2 are those of H2O and H2 molecules separately. In this work, the applied potential was considered by the computational hydrogen electrode method (CHE), with ΔGU = -neU term added to the reaction free energy, where U is the electrode applied potential, e is the transferred charge, and n is the number of proton-electron transferred pairs. pH corrections (ΔGpH) were calculated by kBT×ln10×pH, where kB is the Boltzmann constant and T is the temperature. Reaction free energies were calculated to qualitatively describe the possible reaction pathway for O2/O3 formation. Adding the kBT×ln10×pH term in each elementary step would not affect the main conclusion in this work, and thus the pH effect was not included here.
AIMD simulations were also carried out at the Γ point implemented in the VASP code, with the time step length to be 1 fs and the system temperature maintained at 298 K by using the Nose-Hoover thermostat in the NVT ensemble.[45,46] The solvent condition was also considered by adding water molecules to the vacuum gap. Within the constructed PbO2 (110) surface model, the vacuum gap was predicted to be 10.37Å × 14.31Å × 14.17Å. 20 water molecules were added in the vacuum gap with the density calculated to be 0.28 g/cm3, which was lower than that of the standard value (1.0 g/cm3) due to the large vacuum gap in the constructed slab model. Depending on the slab model mentioned above, more H2O molecules (~70) should be inserted in the vacuum to reach the target density of 1.0 g/cm3. However, the calculations were limited due to the significant computational cost by introducing such large amount of H2O molecules. In this work, the AIMD simulation was applied to only provide qualitative discussions of the AEM/LOM reaction pathway. Thus, the slab containing 20 H2O molecules was used throughout AIMD simulations. For the relative distributions of H2O molecules, van-der Walls interaction was considered when constructing the initial structure for AIMD calculation, with the COMPASS force field used in this process.[47] The structure was first pre-equilibrated using the classical MD calculation before AIMD simulation.
RESULTS AND DISCUSSION
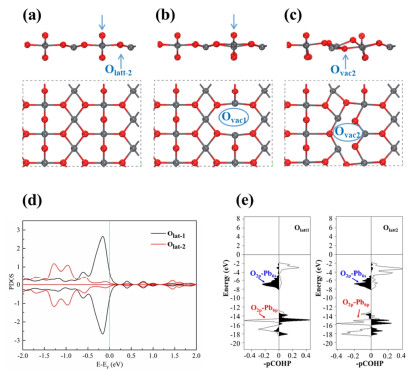
Structure and Stability of Surface Lattice Oxygen Site on Pristine PbO2(110). For the pristine PbO2 (110) surface, two types of lattice oxygen atoms were exposed: the first is the surface lattice oxygen atoms (Olatt-1) with the coordination number of 2, which distribute with the zigzag configuration; and the other type coordinates with 3 oxygen atoms on the exposed surface, as shown in Figure 1(a). Previous work by Wang and coworkers has fully explored the stability or the surface lattice oxygens by calculating the oxygen vacancy formation energies, [33] showing that the oxygen vacancy formation energies were calculated to be -1.23 and -0.59 eV, respectively by removing the Olatt-1 and Olatt-2 from the (110) surface.[33] Despite the over-binding of gaseous O2 molecule and underestimation of O2 energy, [48-54] the surface oxygen vacancy formation still remains thermodynamically favorable by adding the correction values of ~0.80 eV at the PBE level. As a result, the Olatt-1/Olatt-2 site on the (110) surface is likely to encounter the facile diffusion and coupling to generate gaseous O2/O3, leaving behind the Ovac1 and Ovac2 sites on the surface, which would promote the subsequent H2O adsorption and refilling process.
Figure 1
Figure 1.
The optimized structure of (a) pristine PbO2 (110), (b) PbO2 (110) with Ovac1 and (c) PbO2 (110) with Ovac2. The upper and lower figures in each sub-figure are side and top views, respectively. (d) Projected density states (PDOS) for the 2p orbital of the Olatt-1 and Olatt-2 sites on the pristine PbO2 (110) surface. (e) Crystal orbital Hamilton population analysis (COHP) of the Pb-Olatt-1 and Pb-Olatt-2 bonds. Copyright 2022, American Chemical Society.
Furthermore, the PbO2(110)-Ovac2 surface would reconstruct during the structural optimization, as shown in Figure 1(c). The above results demonstrated higher stability of the Olatt-2 site ascribed to the different Pb-O covalency.[33] Therefore, the projected density of states was used to qualitatively describe the Pb-Olatt-1/Olatt-2 covalency, with corresponding results depicted in Figure 1(d). For pristine PbO2 (110) surface, the valence state for Pb was generally predicted to be +4, indicating that four electrons were transferred from Pb to neighboring O sites with the valence electron distribution of 6s05d106p0. The 5d orbital was still fulfilled, resulting in Pb-6s/6p participating in the covalent interactions with O-2p orbital. Crystal orbital Hamilton population analysis (COHP) also demonstrated that the covalent interaction between Pb and the Olatt-1/latt-2 site was dominated by the Pb-6s/6p interaction with the O-2p orbital.
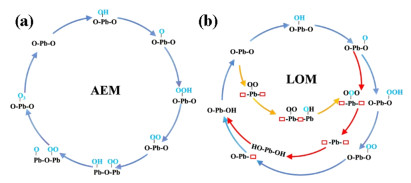
Mechanistic Study of Competitive AEM/LOM Reaction Pathway. Under basic conditions (pH > 7), hydroxide anions (OH-) are oxidized to produce O2/O3 in the OER and EOP reaction on the anode, while H+ protons are reduced to H2 on the cathode. It was worth mentioning that only the half-reaction was investigated to reveal the electrocatalytic mechanism of O2/O3 formation, in which the reaction was initiated by OH- adsorption on the surface to form OH* adsorbate. Detailed reaction intermediates for proposed pathways are shown in Scheme 1. For AEM reaction pathway, the O3 formation consists of six PCET elementary steps and one O2-O coupling step, [55,56] as shown in the following equations:
Scheme 1.
Schematic diagram of the proposed reaction network of (a) AEM pathway and (b) LOM pathway. Adsorbate species originated from H2O are shown in blue font.
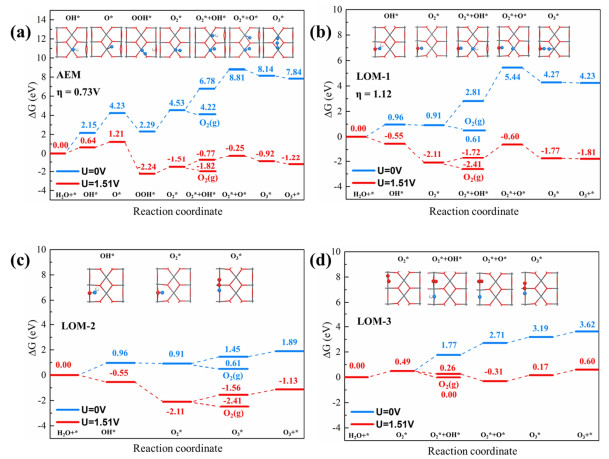
Whereas for LOM mechanism, three possible reaction pathways were proposed (LOM-1, LOM-2, and LOM-3), as shown in blue, red and yellow reaction pathways in Scheme 1 (b), respectively. Within the proposed LOM-1 reaction pathway, the O2* was initially generated from the four-step PCET mechanism. Instead of direct desorption, the O2* intermediate further coupled with the exposed surface lattice oxygen to form O3. In proposed LOM-2 pathway, the adsorbed O* intermediate originated from H2O further coupled with two surface lattice oxygen atoms to form O3, leaving behind two oxygen vacancy sites which are further fulfilled by OH- in the solvent condition. In LOM-3 pathway, the O2* was firstly generated via the surface lattice oxygen diffusion and coupling, which further reacted with the adsorbed OH* to form O3 product. Detailed reaction energies as well as the theoretical over-potential are shown in free energy diagrams in Figure 2.
Figure 2
Figure 2.
DFT calculated free energy diagrams of (a) AEM pathway, (b) LOM-1 pathway, (c) LOM-2 pathway and (d) LOM-3 pathway.
For the AEM reaction pathway, we suppose that the reaction occurred in the alkali solvent condition, initiating with OH- adsorption on the Pb site form OH*. The free energy change was calculated to be 2.15 eV at 298 K and U = 0.00 V. Upon the adsorbed OH* intermediate, the reaction was then followed by two-step electron elementary steps, with O* and OOH* intermediates formed on the surface. Reaction energies were calculated to be 2.08 and -1.94 eV, respectively. Note that we are unable to locate the local-minimum structure of OOH* adsorbate, since the fragment would easily dissociate to form Pb-O2*/Olatt-H* intermediate and the Olatt acted as the Brønsted basic site in this process. In step 4, the OOH* intermediate further reacted with OH- to get the O2* intermediate. Based on calculation, the reaction was also highly endothermic (2.24 eV) under the applied potential of 0.00 V. After forming the O2* intermediate, two competitive reaction pathways accompanied with the O2/O3 formation were proposed. The first is the O2* direct desorption to form gaseous O2, with the reaction energy calculated to be slightly exothermic of -0.31 eV. Alternatively, the O2* was also likely to further react with OH- to form O3* via two elementary reaction steps, with the reaction energies calculated to be 2.25 and 2.03 eV, respectively for the formation of O2*+OH* and O2*+O*. The adsorbed separated O2*+O* intermediates further coupled to generate the target product (O3*), and the calculated reaction energy was -0.67 eV. In the proposed AEM mechanism, the reaction energies of O2* and O2*+OH* were calculated to be comparable to each other. Despite the dissociation of OOH* intermediate and the raised O2* formation energy, O2*+OH* was treated as the potential-determining step (PDS) with the theoretical over-potential predicted (η) to be 0.73 V under the applied potential of 1.51 V.
In LOM-1 pathway, the reaction was initiated by OH- adsorbing on Olatt to form the OlattOH* intermediate. The reaction energy and Olatt-OH* bond length were calculated to be 0.96 eV and 1.42 Å, respectively. The deprotonation step was then followed to generate OlattO* with the reaction energy of -0.05 eV. After forming OlattO* adsorbate, the reaction was then followed by two PCET steps to form the OlattO*+OH* and OlattO*+O* intermediates, with the reaction energies calculated to be 1.90 and 2.63 eV, respectively. The O3* was subsequently generated via OlattO*, O* coupling. It is worth noting that in the LOM-1 pathway, the exposed Olatt and Pb sites play different roles for O3 selective formation: The Olatt attracts the adsorption of OH- and stabilizes the OlattO* intermediate, while the Pb provides additional adsorption and evolution site of OH-. The O2*+O* formation step was predicted to be the PDS and the ηEOP was calculated to be 1.12 V. It should be noted that after forming OlattO* intermediate, the exposed surface lattice oxygen was also likely to diffuse and couple with OlattO* to form OlattOOlatt* (O3*). The reaction energy was calculated to be slightly endothermic of 0.54 eV (LOM-2 in Figure 2(b)). Different from LOM-1 and LOM-2, the oxygen atoms in O2* intermediate were originated from Olatt in LOM-3 reaction pathway. The corresponding free energy diagram is shown in Figure 2(d). The reaction energy of Olatt diffusion and coupling was calculated to be only 0.49 eV, and those following the oxygen evolution to O2*+OH*/O2*+O* to be 1.28 and 0.94 eV, respectively. The O3* was generated via the O2*-O* coupling with a moderate reaction energy of 0.46 eV.
Upon the above results, the OlattOH* formation was predicted to be PDS in the LOM-2 pathway, with the reaction energy calculated to be 0.96 eV, whereas for LOM-3, PDS was predicted to be the OlattOlatt*+OH* formation step, and the calculated reaction energy for this OH- adsorption step was 1.28 eV. However, both the two values are lower than 1.51 eV (the equilibrium potential of EOP). Hence, the theoretical over-potential could not be determined in LOM-2 and LOM-3, also indicating the reaction could occur spontaneously under the condition of 1.51 V.
The selective formation of O2/O3 was mainly ascribed to competitive reaction pathways after the O2* intermediate formation: O2* is directly desorbed to form gaseous O2, and O2* further reacts and couples with OH- or surface lattice oxygen (Olatt) to form O3*. For all reaction pathways involved in O2/O3 formation, the O2 formation was predicted to be thermodynamically more favorable with the condition of U = 0.0 and 1.51 V. However, for elementary steps coupled with proton and electron transfer, the applied external potential has significant impact on the reaction free energy due to the ΔGU term introduced. For AEM reaction path, the energy difference of OH- adsorption on O2* pre-occupied surface to OH*+O2* and O2* direct desorption was decreased from 2.56 to 1.05 eV with the U value of 0.00 and 1.51 V, respectively; whereas for LOM-1 pathway, the energy difference of the above processes drops from 2.20 to 0.69 eV with the increase of U value. Therefore, the crossover point for O2/O3 selectivity inversion was predictable when increasing the applied potential. Furthermore, the above results show that the reaction free energies from PCET steps are more sensitive toward the U value upon the CHE method, which would result in the LOM reaction pathways more favorable at the low potential range, while the AEM reaction mechanism is likely to dominate the OER/EOP reaction at the high potential range. In addition to the applied potential effect on the reaction energies and product selectivity in OER/EOP process, the adsorption configurations for each reaction intermediate would also be affected by the applied potential. Previous work by Wang and coworkers has fully investigated the potential-dependent CO2 reduction reaction over the single atom catalysts, with the results showing that CO2 chemisorption structure is significantly affected by the applied potential: CO2 molecule charged to form the linear CO2-, with transition states and final states also polarized and stabilized due to charge redistribution under the potential-dependent solvation condition.[57] The ΔG~U linear relationships were also obtained, which can be further used to determine the onset potential of CO2RR, the potential at the maximal CO Faraday efficiency (FE), and the potential at FECO = FEH2 (Ucross).[57] This work gives a comprehensive understanding of O2/O3 selective formation via multiple reaction mechanisms, and further investigations are required for the explicit understanding for applied potential effect on adsorption configurations and reaction energies.
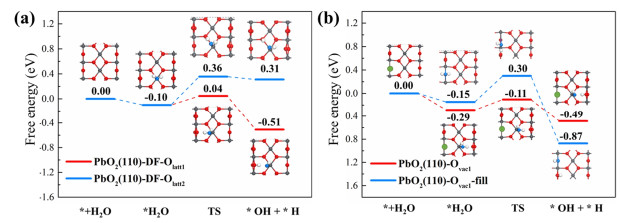
H2O Adsorption and Dissociation on PbO2 (110) Surface with the Effect of Oxygen Vacancy. Depending on the above results, the proposed LOM-2/LOM-3 pathway was more favorable than the AEM reaction, indicating that the surface lattice oxygen would easily diffuse and desorb from the surface. Hence, it is possible to result in the PbO2 (110) surface structure collapsion. However, experimental results revealed the considerable stability of PbO2 nanorod catalyst exposing (110) and (101) surface.[22] This is most likely attributed to the underlying H2O adsorption/dissociation process, which fulfills the produced oxygen vacancy site under real reaction conditions. Therefore, H2O splitting on pristine and oxygen vacancy-rich PbO2 (110) surface was studied to reveal the surface oxygen vacancy refilling mechanism, with corresponding results depicted in Figure 3.
Figure 3
Figure 3.
Free energy diagram (eV) for H2O splitting on (a) pristine PbO2 (110) surface and (b) PbO2 (110) surface with Ovac1.
For pristine PbO2 (110) surface, the H2O splitting follows the acid-base interaction mechanism: The OH-/H+ yielded from H2O dissociation binds to the Lewis acid (Pb) and Brønsted base (Olatt‑1/Olatt‑2) sites, respectively. The reactivity of different Brønsted base sites including Olatt‑1 and Olatt‑2 was systematically investigated. Within Pb-OH/H-Olatt‑1 configuration, the reaction energy of H2O splitting was calculated to be -0.41 eV, much lower than that of the Pb-OH/H-Olatt‑2 intermediate (0.41 eV). The free energy barrier of H2O splitting on the Olatt‑1 site was also calculated to be 0.32 eV lower than that on Olatt‑2. The above results revealed that the exposed Olatt‑1 site exhibited a higher reactivity than the Olatt‑2 one. For the calculated transition states, only one imaginary frequency was predicted.
Considering the higher reactivity of Olatt‑1 site and lower Ovac1 formation energy, Ovac1 formation was predicted to be more thermodynamically favorable than the Ovac1 formation process. Reactivity of H2O dissociation on the PbO2 (110) surface was subsequently investigated. The results show that H2O adsorption and dissociation on the Pb site were also calculated to be more favorable with the adsorption and dissociation energy calculated to be -0.29 and -0.20 eV, respectively. The free energy barrier of this step was calculated to be only 0.18 eV. When considering the reaction pathway of H2O fulfilling the Ovac1 site, the adsorption and dissociation energies were calculated to be -0.15 and -0.72 eV, respectively. The calculated activation energy was slightly lower than that in the reaction pathway of dissociation on the Pb site, which was calculated to be 0.15 eV. Although the potential energy surface of H2O splitting via the acid-base interaction was lower than that of the direct H2O fulfilling pathway, the energy barrier for the direct H2O fulfilling pathway was slightly lower (0.03 eV). Additionally, the OH fragment in the Pb-OH/H-Olatt‑1 intermediate also likely migrated from the Pb site to Ovac1 due to the energy difference of 0.38 eV.
Considering the facile splitting of H2O on the PbO2 (110) surface with and without oxygen vacancy sites, reaction equilibrium of H2O dissociation to OH- and H+ was also likely to be promoted, which further enhanced the evolution of OH- to OH*, as shown in equation 1. Additionally, basic hydroxyl adsorption on Pb site (OH*) over the pristine PbO2 (110) surface was unlikely to reach the equilibrium coverage since the OH* fragment is also possible to diffuse on the surfaces via the active proton exchange with the undissociated adsorption water (H2O∗). Previous work by Xia et al also revealed the OH* migration on the monoclinic ZrO2($ \overline 1 $11) surface by AIMD simulations, and the equilibrium coverage of OH* and H2O* under different calcination temperatures was predicted.[58]
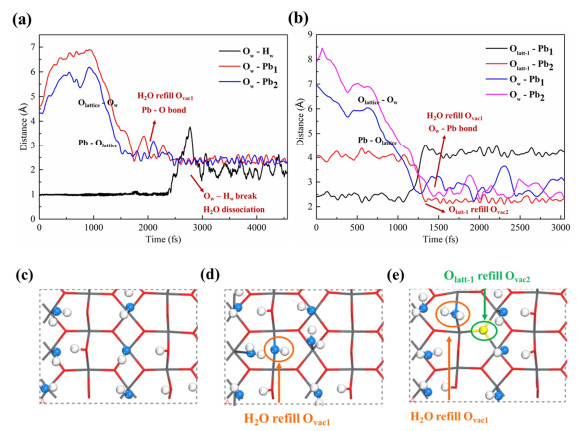
AIMD Simulation of Water on the PbO2 (110) Surface. AIMD simulations were further carried out to qualitatively reveal the oxygen refilling process on PbO2 (110) with oxygen vacancies, which aims to explain the durability of β-PbO2 NRs catalyst under real reaction conditions.[22] The AIMD simulations were employed at 298 K to illustrate how the oxygen vacancy affects the H2O splitting on PbO2 (110). The effects of Ovac1 and Ovac2 on the H2O splitting have also been investigated, with corresponding results presented in Figure 4 (a) and (b). Over the pristine PbO2 (110) surface without oxygen vacancies, H2O molecules were observed to dissociate and adsorb on the surface with the two-fold surface lattice oxygen site (Olatt-1) protonated to HOlatt-1*, and the basic hydroxyls (OH*) chemisorbed on the Pb sites. The corresponding structure is shown in Figure 4 (c).
Figure 4
Figure 4.
Ab initio molecular dynamics (AIMD) modelling for the dissociation of H2O on PbO2 (110) surface. (a) Bond distance plots of Pb-Ow and Ow-Hw on PbO2 (110) surface with Ovac1, (b) bond distance plots of Pb-Ow and Olatt-1-Pb on PbO2 (110) surface with Ovac2. Selected snapshots of the AIMD trajectory for water dissociation at 298 K on (c) PbO2 (110)-DF surface, (d) PbO2 (110)-Ovac1 surface and (e) PbO2 (110)-Ovac2 surface.
For PbO2 (110) surface pre-occupied by the Ovac1 site, comparable results were yielded in the initial stage for H2O adsorption and dissociation, and oxygen vacancy refilling process was also found. Bond distance plots between oxygen atom from H2O (Ow) and surface Pb sites show that covalent Pb-Ow bond was generated at the time scale of 1.7-4.5 ps, and the Pb-Ow bond length was predicted to be -2.30 Å. For PbO2 (110) surface pre-occupied by the Ovac2 site, the exposed Olatt-1 site was easy to diffuse under aqueous condition at 298 K, with the Ovac2 site fulfilled, leaving behind the Ovac1 site on the surface at the time scale of 1.0-1.3 ps. The OH* refilling the Ovac1 site was also observed at the time scale of 0.7-1.5 ps. The above results qualitatively demonstrated the H2O dissociation and diffusion on the PbO2 (110) surface with and without oxygen vacancies, which also explained the experimental durability of β-PbO2 NR catalyst exposing the (110) and (101) surfaces: surface oxygen vacancy sites generated by LOM mechanism are possible to be refilled by OH* from H2O.
CONCLUSIONS
The EOP reactivity on PbO2 (110) surface was extensively investigated, with the detailed reaction network proposed (AEM, LOM-1, LOM-2, and LOM-3). The results show that for O3 formation, the LOM-2 reaction pathway was the most thermodynamically favorable, and the first OH* formation step was predicted to be the potential-determining step. Considering the facile diffusion of surface lattice oxygen atoms and formation of oxygen vacancies of the PbO2 (110) surface, the catalyst was considered to be regenerated by H2O adsorption and splitting, resulting in surface oxygen vacancy refilling, which also ensured the stability of the PbO2 (110) surface structure. DFT calculations proved that the H2O dissociation was dominated by the acid-base interaction mechanism for the PbO2 (110) surface with and without oxygen vacancy. Pb-OH fragment on the PbO2 (110)-Ovac1 surface was also likely to migrate to fulfill the Ovac1 due to the energy difference of 0.38 eV. By comparing the reaction energy and energy barrier for H2O adsorption/dissociation between pristine PbO2 (110) and PbO2 (110)-Ovac1, it is also possible to conclude that the introduction of surface oxygen vacancy (Ovac1) could promote the H2O splitting, which was further verified by AIMD simulations. The above results provided in-depth understanding of the O2/O3 selective formation on the PbO2 (110) electrode surface.
ACKNOWLEDGEMENTS:
This work was financially supported by the National Key R & D Program of China (2021YFA1500900) and National Natural Science Foundation of China (21625604, 21878272, 22141001).
W. W. Li contributed in writing the manuscript and performing the DFT calculation. S. B. Wang and J. G. Wang designed the research and wrote the paper. All authors commented on and revised the manuscript.
AUTHOR CONTRIBUTION COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information is available for this paper at http://manu30.magtech.com.cn/jghx/EN/10.14102/j.cnki.0254-5861.2022-0153 For submission:https://www.editorialmanager.com/cjschem ADDITIONAL INFORMATION
[1]
Kasprzyk-Hordern, B.; Ziółek, M.; Nawrocki, J. Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment. Appl. Catal., B: Environ.2003, 46, 639-669. doi: 10.1016/S0926-3373(03)00326-6
[2]
Cremer, D.; Crehuet, R.; Anglada, J. The ozonolysis of acetylenea quantum chemical investigation. J. Am. Chem. Soc.2001, 123, 6127-6141. doi: 10.1021/ja010166f
[3]
Criegee, R. Mechanism of ozonolysis. Angew. Chem. Int. Ed.1975, 14, 745-752. doi: 10.1002/anie.197507451
[4]
Smith, G. D.; Woods, E.; DeForest, C. L.; Baer, T.; Miller, R. E. Reactive uptake of ozone by oleic acid aerosol particles: application of single-particle mass spectrometry to heterogeneous reaction kinetics. J. Phys. Chem. A2002, 106, 8085-8095. doi: 10.1021/jp020527t
[5]
Khadre, M. A.; Yousef, A. E.; Kim, J. G. Microbiological aspects of ozone applications in food: a review. J. Food Sci.2001, 66, 1242-1252. doi: 10.1111/j.1365-2621.2001.tb15196.x
[6]
Christensen, P. A.; Yonar, T.; Zakaria, K. The electrochemical generation of ozone: a review. Ozone: Sci. Eng.2013, 35, 149-167. doi: 10.1080/01919512.2013.761564
[7]
Lee, Y.; Suntivich, J.; May, K. J.; Perry, E. E.; Shao-Horn, Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions. J. Phys. Chem. Lett.2012, 3, 399-404. doi: 10.1021/jz2016507
[8]
Reier, T.; Oezaslan, M.; Strasser, P. Electrocatalytic oxygen evolution reaction (OER) on Ru, Ir, and Pt catalysts: a comparative study of nanoparticles and bulk materials. ACS Catal.2012, 2, 1765-1772. doi: 10.1021/cs3003098
[9]
Seitz Linsey, C.; Dickens Colin, F.; Nishio, K.; Hikita, Y.; Montoya, J.; Doyle, A.; Kirk, C.; Vojvodic, A.; Hwang Harold, Y.; Norskov Jens, K.; Jaramillo Thomas, F. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science2016, 353, 1011-1014. doi: 10.1126/science.aaf5050
[10]
Yang, L.; Yu, G.; Ai, X.; Yan, W.; Duan, H.; Chen, W.; Li, X.; Wang, T.; Zhang, C.; Huang, X.; Chen, J. S.; Zou, X. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers. Nat. Commun.2018, 9, 5236. doi: 10.1038/s41467-018-07678-w
[11]
Zhang, C.; Xu, Y.; Lu, P.; Zhang, X.; Xu, F.; Shi, J. Capillary effectenabled water electrolysis for enhanced electrochemical ozone production by using bulk porous electrode. J. Am. Chem. Soc.2017, 139, 16620-16629. doi: 10.1021/jacs.7b07705
[12]
Awad, M. I.; Saleh, M. M.; Ohsaka, T. Ozone electrogeneration on Pt-loaded reticulated vitreous carbon using flooded and flow-through assembly. J. Electrochem. Soc.2006, 153, D207-D212. doi: 10.1149/1.2358837
[13]
Da Silva, L. M.; De Faria, L. A.; Boodts, J. F. C. Electrochemical ozone production: influence of the supporting electrolyte on kinetics and current efficiency. Electrochim. Acta2003, 48, 699-709. doi: 10.1016/S0013-4686(02)00739-9
[14]
Cheng, S. -A.; Chan, K. -Y. Electrolytic generation of ozone on an antimony-doped tin dioxide coated electrode. Electrochem. Solid-State Lett.2004, 7, D4-D6. doi: 10.1149/1.1645753
[15]
Shekarchizade, H.; Amini, M. K. Effect of elemental composition on the structure, electrochemical properties, and ozone production activity of Ti/SnO2-Sb-Ni electrodes prepared by thermal pyrolysis method. Int. J. Electrochem.2011, 2011, 17580-17590.
[16]
Arihara, K.; Terashima, C.; Fujishima, A. Application of freestanding perforated diamond electrodes for efficient ozone-water production. Electrochem. Solid-State Lett.2006, 9, D17-D20. doi: 10.1149/1.2206009
[17]
Kraft, A.; Stadelmann, M.; Wünsche, M.; Blaschke, M. Electrochemical ozone production using diamond anodes and a solid polymer electrolyte. Electrochem. Commun.2006, 8, 883-886. doi: 10.1016/j.elecom.2006.02.013
[18]
Fu, H. -C.; Varadhan, P.; Tsai, M. -L.; Li, W.; Ding, Q.; Lin, C. -H.; Bonifazi, M.; Fratalocchi, A.; Jin, S.; He, J. -H. Improved performance and stability of photoelectrochemical water-splitting Si system using a bifacial design to decouple light harvesting and electrocatalysis. Nano Energy2020, 70, 104478. doi: 10.1016/j.nanoen.2020.104478
[19]
Lin, S.; Huang, H.; Ma, T.; Zhang, Y. Photocatalytic oxygen evolution from water splitting. Adv. Sci.2021, 8, 2002458. doi: 10.1002/advs.202002458
[20]
Zhang, B.; Wang, L.; Cao, Z.; Kozlov, S. M.; García de Arquer, F. P.; Dinh, C. T.; Li, J.; Wang, Z.; Zheng, X.; Zhang, L.; Wen, Y.; Voznyy, O.; Comin, R.; De Luna, P.; Regier, T.; Bi, W.; Alp, E. E.; Pao, C. -W.; Zheng, L.; Hu, Y.; Ji, Y.; Li, Y.; Zhang, Y.; Cavallo, L.; Peng, H.; Sargent, E. H. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal.2020, 3, 985-992. doi: 10.1038/s41929-020-00525-6
[21]
Devilliers, D.; Dinh Thi, M. T.; Mahé, E.; Le Xuan, Q. Cr(Ⅲ) oxidation with lead dioxide-based anodes. Electrochim. Acta2003, 48, 4301-4309. doi: 10.1016/j.electacta.2003.07.005
Jahangiri, S.; Mosey, N. J. Computational investigation of the oxygen evolution reaction catalyzed by nickel (oxy)hydroxide complexes. J. Phys. Chem. C2018, 122, 25785-25795. doi: 10.1021/acs.jpcc.8b06614
[24]
Liu, T.; Feng, Z.; Li, Q.; Yang, J.; Li, C.; Dupuis, M. Role of oxygen vacancies on oxygen evolution reaction activity: β-Ga2O3 as a case study. Chem. Mater.2018, 30, 7714-7726. doi: 10.1021/acs.chemmater.8b03015
[25]
Man, I. C.; Su, H. -Y.; Calle-Vallejo, F.; Hansen, H. A.; Martínez, J. I.; Inoglu, N. G.; Kitchin, J.; Jaramillo, T. F.; Nørskov, J. K.; Rossmeisl, J. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem.2011, 3, 1159-1165. doi: 10.1002/cctc.201000397
[26]
Murdachaew, G.; Laasonen, K. Oxygen evolution reaction on nitrogen-doped defective carbon nanotubes and graphene. J. Phys. Chem. C2018, 122, 25882-25892. doi: 10.1021/acs.jpcc.8b08519
[27]
Rossmeisl, J.; Logadottir, A.; Nørskov, J. K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys.2005, 319, 178-184. doi: 10.1016/j.chemphys.2005.05.038
[28]
Valdés, Á.; Qu, Z. W.; Kroes, G. J.; Rossmeisl, J.; Nørskov, J. K. Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C2008, 112, 9872-9879. doi: 10.1021/jp711929d
[29]
Wei, R.; Bu, X.; Gao, W.; Villaos, R. A. B.; Macam, G.; Huang, Z. -Q.; Lan, C.; Chuang, F. -C.; Qu, Y.; Ho, J. C. Engineering surface structure of spinel oxides via high-valent vanadium doping for remarkably enhanced electrocatalytic oxygen evolution reaction. ACS Appl. Mater. Int.2019, 11, 33012-33021. doi: 10.1021/acsami.9b10868
[30]
Grimaud, A.; Hong, W. T.; Shao-Horn, Y.; Tarascon, J. M. Anionic redox processes for electrochemical devices. Nat. Mater.2016, 15, 121-126. doi: 10.1038/nmat4551
[31]
Mefford, J. T.; Rong, X.; Abakumov, A. M.; Hardin, W. G.; Dai, S.; Kolpak, A. M.; Johnston, K. P.; Stevenson, K. J. Water electrolysis on La1-xSrxCoO3-δ perovskite electrocatalysts. Nat. Commun.2016, 7, 11053. doi: 10.1038/ncomms11053
[32]
Suntivich, J.; May Kevin, J.; Gasteiger Hubert, A.; Goodenough John, B.; Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science2011, 334, 1383-1385. doi: 10.1126/science.1212858
[33]
Li, W.; Feng, G.; Wang, S.; Liu, J.; Zhong, X.; Yao, Z.; Deng, S.; Wang, J. Lattice oxygen of PbO2 (101) consuming and refilling via electrochemical ozone production and H2O dissociation. J. Phys. Chem. C2022, 126, 8627-8636. doi: 10.1021/acs.jpcc.2c00725
[34]
Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci.1996, 6, 15-50. doi: 10.1016/0927-0256(96)00008-0
[35]
Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B1996, 54, 11169-11186. doi: 10.1103/PhysRevB.54.11169
[36]
Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett.1996, 77, 3865-3868. doi: 10.1103/PhysRevLett.77.3865
[37]
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B1994, 50, 17953-17979. doi: 10.1103/PhysRevB.50.17953
[38]
Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B1999, 59, 1758-1775.
[39]
Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys.2000, 113, 9978-9985. doi: 10.1063/1.1323224
[40]
Henkelman, G.; Uberuaga, B. P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys.2000, 113, 9901-9904. doi: 10.1063/1.1329672
[41]
Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys.1999, 111, 7010-7022. doi: 10.1063/1.480097
[42]
Heyden, A.; Bell, A. T.; Keil, F. J. Efficient methods for finding transition states in chemical reactions: comparison of improved dimer method and partitioned rational function optimization method. J. Chem. Phys.2005, 123, 224101. doi: 10.1063/1.2104507
[43]
Kästner, J.; Sherwood, P. Superlinearly converging dimer method for transition state search. J. Chem. Phys.2008, 128, 014106. doi: 10.1063/1.2815812
[44]
Xiao, P.; Sheppard, D.; Rogal, J.; Henkelman, G. Solid-state dimer method for calculating solid-solid phase transitions. J. Chem. Phys.2014, 140, 174104. doi: 10.1063/1.4873437
[45]
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A1985, 31, 1695-1697. doi: 10.1103/PhysRevA.31.1695
[46]
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys.1984, 81, 511-519. doi: 10.1063/1.447334
[47]
Sun, H. COMPASS: an ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B1998, 102, 7338-7364. doi: 10.1021/jp980939v
[48]
Chevrier, V. L.; Ong, S. P.; Armiento, R.; Chan, M. K. Y.; Ceder, G. Hybrid density functional calculations of redox potentials and formation energies of transition metal compounds. Phys. Rev. B2010, 82, 075122. doi: 10.1103/PhysRevB.82.075122
[49]
Giannozzi, P.; Car, R.; Scoles, G. Oxygen adsorption on graphite and nanotubes. J. Chem. Phys.2003, 118, 1003-1006. doi: 10.1063/1.1536636
[50]
Hammer, B.; Hansen, L. B.; Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B1999, 59, 7413-7421. doi: 10.1103/PhysRevB.59.7413
[51]
Mutter, D.; Urban, D. F.; Elsässer, C. Determination of formation energies and phase diagrams of transition metal oxides with DFT+U. Materials2020, 13, 4303. doi: 10.3390/ma13194303
[52]
Pople, J. A.; Head-Gordon, M.; Fox, D. J.; Raghavachari, K.; Curtiss, L. A. Gaussian-1 theory: a general procedure for prediction of molecular energies. J. Chem. Phys.1989, 90, 5622-5629. doi: 10.1063/1.456415
[53]
Sai Gautam, G.; Carter, E. A. Evaluating transition metal oxides within DFT-SCAN and SCAN+U frameworks for solar thermochemical applications. Phys. Rev. Mater.2018, 2, 095401. doi: 10.1103/PhysRevMaterials.2.095401
[54]
Yoo, J. S.; Rong, X.; Liu, Y.; Kolpak, A. M. Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites. ACS. Catal.2018, 8, 4628-4636. doi: 10.1021/acscatal.8b00612
[55]
Gibson, G.; Morgan, A.; Hu, P.; Lin, W. -F. New insights into electrocatalytic ozone generation via splitting of water over PbO2 electrode: a DFT study. Chem. Phys. Lett.2016, 654, 46-51. doi: 10.1016/j.cplett.2016.04.078
[56]
Gu, Y.; Wang, S.; Shi, H.; Yang, J.; Li, S.; Zheng, H.; Jiang, W.; Liu, J.; Zhong, X.; Wang, J. Atomic Pt embedded in BNC nanotubes for enhanced electrochemical ozone production via an oxygen intermediate-rich local environment. ACS Catal.2021, 11, 5438-5451. doi: 10.1021/acscatal.1c00413
[57]
Cao, H.; Zhang, Z.; Chen, J. -W.; Wang, Y. -G. Potential-dependent free energy relationship in interpreting the electrochemical performance of CO2 reduction on single atom catalysts. ACS Catal.2022, 12, 6606-6617. doi: 10.1021/acscatal.2c01470
[58]
Xia, G. J.; Wang, Y. G. Dynamic simulation on surface hydration and dehydration of monoclinic zirconia. Chin. J. Chem. Phys.2022, 35, 629-638. doi: 10.1063/1674-0068/cjcp2204062
Figure 1
The optimized structure of (a) pristine PbO2 (110), (b) PbO2 (110) with Ovac1 and (c) PbO2 (110) with Ovac2. The upper and lower figures in each sub-figure are side and top views, respectively. (d) Projected density states (PDOS) for the 2p orbital of the Olatt-1 and Olatt-2 sites on the pristine PbO2 (110) surface. (e) Crystal orbital Hamilton population analysis (COHP) of the Pb-Olatt-1 and Pb-Olatt-2 bonds. Copyright 2022, American Chemical Society.
Scheme 1
Schematic diagram of the proposed reaction network of (a) AEM pathway and (b) LOM pathway. Adsorbate species originated from H2O are shown in blue font.
Figure 4
Ab initio molecular dynamics (AIMD) modelling for the dissociation of H2O on PbO2 (110) surface. (a) Bond distance plots of Pb-Ow and Ow-Hw on PbO2 (110) surface with Ovac1, (b) bond distance plots of Pb-Ow and Olatt-1-Pb on PbO2 (110) surface with Ovac2. Selected snapshots of the AIMD trajectory for water dissociation at 298 K on (c) PbO2 (110)-DF surface, (d) PbO2 (110)-Ovac1 surface and (e) PbO2 (110)-Ovac2 surface.

 DownLoad:
DownLoad:

 下载:
下载:






