-
[1]
Zhou Q L.. Privileged Chiral Ligands and Catalysts. Wiley-VCH, 2011.
-
[2]
Trost B M, Crawley M L.. Chem. Rev., 2003, 103(8):2921-2944. doi: 10.1021/cr020027w
-
[3]
Shibasaki M, Kanai M.. Chem. Rev., 2008, 108(8):2853-2873. doi: 10.1021/cr078340r
-
[4]
Zhang Z F, Butt N A, Zhang W B.. Chem. Rev., 2016, 116(23):14769-14827. doi: 10.1021/acs.chemrev.6b00564
-
[5]
Zheng K, Liu X H, Feng X M.. Chem. Rev., 2018, 118(16):7586-7656. doi: 10.1021/acs.chemrev.7b00692
-
[6]
Cheng Q, Tu H F, Zheng C, et al.. Chem. Rev., 2019, 119(3):1855-1969. doi: 10.1021/acs.chemrev.8b00506
-
[7]
Yang D X, Wang L Q, Li D, et al.. Chem, 2019, 5(5):1108-1166. doi: 10.1016/j.chempr.2019.02.002
-
[8]
Pellissier H.. Org. Biomol. Chem., 2017, 15:4750-4782. doi: 10.1039/C7OB00903H
-
[9]
Yoshino T, Morimoto H, Lu G, et al.. J. Am. Chem. Soc., 2009, 131(47):17082-17083. doi: 10.1021/ja908571w
-
[10]
Lu G, Yoshino T, Morimoto H, et al.. Angew. Chem. Int. Ed., 2011, 50(19):4382-4385. doi: 10.1002/anie.201101034
-
[11]
Hatano M, Horibe T, Ishihara K, et al.. Org. Lett., 2010, 12(15):3502-3505. doi: 10.1021/ol101353r
-
[12]
Wang L Q, Yang D X, Li D, et al.. Org. Lett., 2015, 17(12):3004-3007. doi: 10.1021/acs.orglett.5b01291
-
[13]
Wang L Q, Li D, Yang D X, et al.. Chem. Asian. J., 2016, 11(5):691-695. doi: 10.1002/asia.201501369
-
[14]
Li D, Wang L Q, Yang D X, et al.. Catal ACS., 2015, 5(12):7432-7436. doi: 10.1021/acscatal.5b02177
-
[15]
Li D, Wang Y J, Wang L Q, et al.. Chem. Commun., 2016, 52(62):9640-9643. doi: 10.1039/C6CC02877B
-
[16]
Lin L, Zhang J L, Ma X J, et al.. Org. Lett., 2011, 13(24):6410-6413. doi: 10.1021/ol202713f
-
[17]
Yang D X, Wang L Q, Han F X, et al.. Angew. Chem. Int. Ed., 2013, 52(26):6739-6742. doi: 10.1002/anie.201301146
-
[18]
Du H F, Zhang X, Wang Z, et al.. Eur. J. Org. Chem., 2008, (13):2248-2254.
-
[19]
Bao H L, Wu J, Li H J, et al.. Eur. J. Org. Chem., 2010, (35):6722-6726.
-
[20]
Li D, Wang K Z, Wang L Q, et al.. Org. Lett., 2017, 19(12):3211-3214. doi: 10.1021/acs.orglett.7b01333
-
[21]
Zhang J L, Liu X H, Wang R.. Chem. Eur. J., 2014, 20(17):4911-4915. doi: 10.1002/chem.201304835
-
[22]
Fujisawa T, Ichiyanagi T, Shimizu M.. Tetrahed. Lett., 1995, 36(28):5031-5034. doi: 10.1016/00404-0399(50)0914X-
-
[23]
Hatano M, Horibe T, Ishihara K.. Angew. Chem. Int. Ed., 2013, 52(17):4549-4553. doi: 10.1002/anie.201300938
-
[24]
Hatano M, Horibe T, Yamashita K, et al.. Asian. J. Org. Chem., 2013, 2(11):952-956. doi: 10.1002/ajoc.201300190
-
[25]
Wang L Q, Yang D X, Li D, et al.. Angew. Chem. Int. Ed., 2018, 57(29):9088-9092. doi: 10.1002/anie.201804177
-
[26]
Hatano M, Nishikawa K, Ishihara K.. J. Am. Chem. Soc., 2017, 139(25):8424-8427. doi: 10.1021/jacs.7b04795
-
[27]
Evans D A, Nelson S G.. J. Am. Chem. Soc. 1997, 119(27):6452-6453.
-
[28]
Elston C L, Jackson R F W, Mac Donald S J F, et al.. Angew. Chem. Int. Ed., 1997, 36(4):410-412. doi: 10.1002/anie.199704101
-
[29]
Jacques O, Richardsb S J, Jackson R F W.. Chem. Commun., 2001, 24:2712-2713.
-
[30]
Noyori R, Suga S, Kawai K, et al.. Pure Appl. Chem., 1988, 60(11):1597-1606. doi: 10.1351/pac198860111597
-
[31]
Yong K H, Taylor N J, Chong J M.. Org. Lett., 2002, 4(23):3553-3556.
-
[32]
Yong K H, Chong J M.. Org. Lett., 2002, 4(23):4139-4142. doi: 10.1021/ol026901p
-
[33]
Anderson J D, García G P G, Hayes D, et al.. Tetrahed. Lett., 2001, 42:7111-7114. doi: 10.1016/S0040-4039(01)01465-4
-
[34]
Henderson K W, Kerr W J, Moir J H.. Chem. Commun., 2000, 6:479-480.
-
[35]
Henderson K W, Kerr W J, Moir J H.. Tetrahedron, 2002, 58:4573-4587. doi: 10.1016/S0040-4020(02)00364-2
-
[36]
Carswell E L, Hayes D, Henderson K W, et al.. Synlett, 2003, 34:1017-1021.
-
[37]
Carswell E L, Kerr W J, McArthur D, et al.. Tetrahedron, 2014, 70:7344-7349. doi: 10.1016/j.tet.2014.06.052
-
[38]
Yang D X, Wang L Q, Han F X, et al.. Angew. Chem. Int. Ed., 2015, 54(7):2185-2189. doi: 10.1002/anie.201410257
-
[39]
Wang L Q, Yang D X, Li D, et al Adv.. Synth. Catal., 2018, 360(23):4491-4496. doi: 10.1002/adsc.201801041
-
[40]
Yang D X, Wang L Q, Kai M, et al.. Angew. Chem. Int. Ed., 2015, 54(33):9523-9527. doi: 10.1002/anie.201503056
-
[41]
Wang L Q, Yang D X, Li D, et al.. Chem. Eur. J., 2016, 22(25):8483-8487. doi: 10.1002/chem.201601399
-
[42]
Galliford C V, Scheidt K A.. Angew. Chem. Int. Ed., 2007, 46(46):8748-8758. doi: 10.1002/anie.200701342
-
[43]
Yu J, Shi F, Gong L.. Acc. Chem. Res., 2011, 44(11):1156-1171. doi: 10.1021/ar2000343
-
[44]
Dalpozzo R, Bartoli G, Bencivenni G.. Chem. Soc. Rev., 2012, 41(21):7247-7290. doi: 10.1039/c2cs35100e
-
[45]
Kotha S, Panguluri N R, Ali R.. Eur. J. Org. Chem., 2017, (36):5316-5342.
-
[46]
Wang L Q, Yang D X, Li D, et al.. Org. Lett., 2015, 17(17):4260-4263. doi: 10.1021/acs.orglett.5b02052
-
[47]
Wang K Z, Wang L Q, Liu X H, et al.. Org. Lett., 2017, 19(16):4351-4354. doi: 10.1021/acs.orglett.7b02044
-
[48]
Li D, Yang D X, Wang L Q, et al.. Chem. Eur. J., 2016, 22(48):17141-17144. doi: 10.1002/chem.201603898
-
[49]
Li D, Wang L Q, Zhu H Y, et al.. Org. Lett., 2019, 21(12):4717-4720. doi: 10.1021/acs.orglett.9b01599
-
[50]
Zhang X M, Emge T J, Hultzsch K C.. Angew. Chem. Int. Ed., 2012, 51(2):394-398. doi: 10.1002/anie.201105079
-
[51]
Zhang M, Tobisch S, Hultzsch K C.. Chem. Eur. J., 2015, 21(21):7841-7857. doi: 10.1002/chem.201406468
-
[52]
Jackowski O, Alexakis A.. Angew. Chem. Int. Ed., 2010, 49(19):3346-3350. doi: 10.1002/anie.201000577
-
[53]
Grassi D, Alexakis A.. Angew. Chem. Int. Ed., 2013, 52(51):13642-13646. doi: 10.1002/anie.201307591
-
[54]
Grassi D, Dolka C, Jackowski O, et al.. Chem. Eur. J., 2013, 19(4):1466-1475. doi: 10.1002/chem.201202318
-
[55]
Yang D X, Wang L Q, Han F X, et al.. Chem. Eur. J., 2014, 20(50):16478-16483. doi: 10.1002/chem.201404354
-
[56]
Wang L Q, Yang D X, Han F X, et al.. Org. Lett., 2015, 17(2):176-179. doi: 10.1021/ol503455r
-
[57]
Yang D X, Wang L Q, Han F X, et al.. Chem. Eur. J., 2014, 20(28):8584-8588. doi: 10.1002/chem.201403183
-
[58]
Li D, Wang L Q, Yang Y L.. Adv. Synth. Catal., 2019, 361(16):3744-3750. doi: 10.1002/adsc.201900481

 下载:
下载:
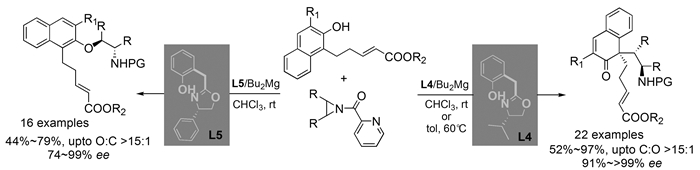
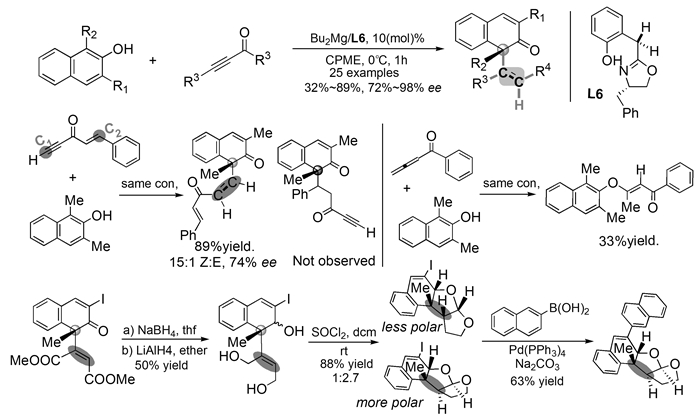
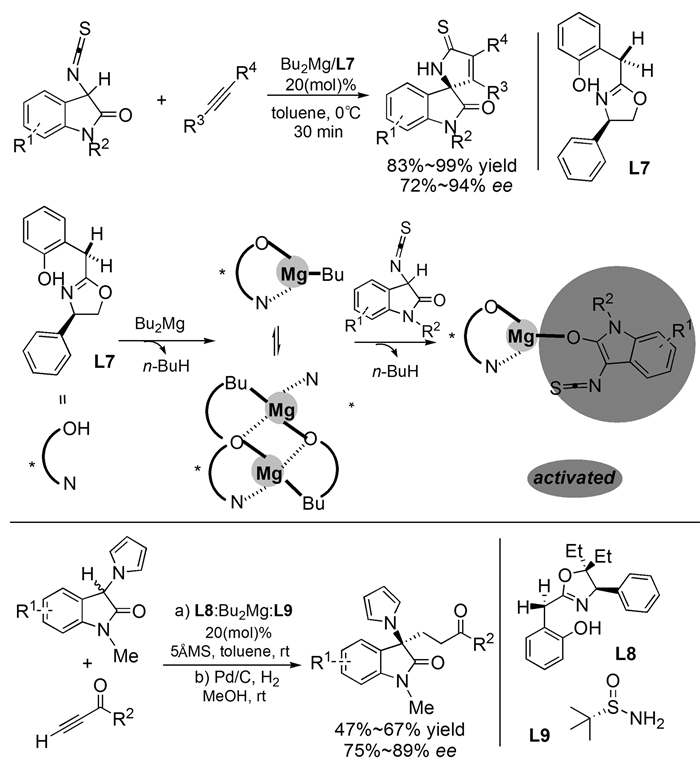
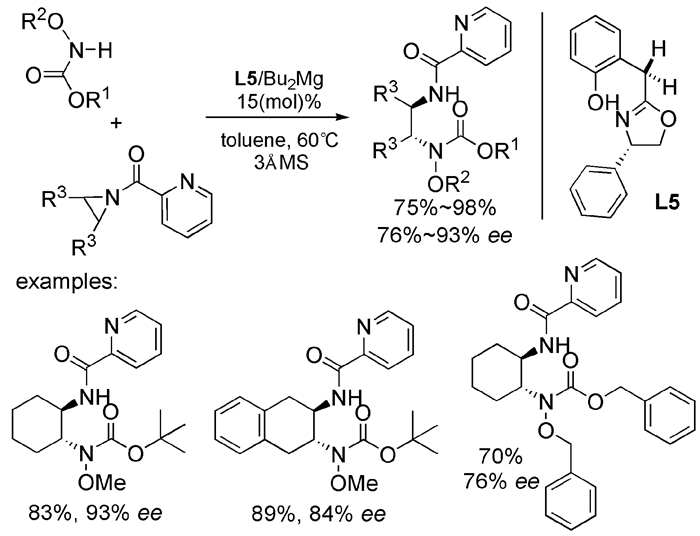

 下载:
下载:



















