School of Physics, Henan Normal University, Xinxiang 453007, China
b.
Department of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Tsinghua University, Beijing 100084, China
c.
Department of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China
d.
School of Chemistry, Minhaj University, Lahore 54000, Pakistan
e.
Department of Chemistry, Mirpur University of Science and Technology (MUST), Mirpur, AJK 10250, Pakistan
f.
College of Engineering, King Saud University, PO Box 800, Riyadh 11421, Saudi Arabia
g.
Chemical Engineering Department, College of Engineering, King Saud University, PO Box 800, Riyadh 11421, Saudi Arabia
h.
Department of Chemistry-Ångström, Uppsala University, Box 538, Uppsala 75121, Sweden
i.
Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
* Corresponding author. ** Corresponding author at: Department of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education
Received Date:
18 January 2022 Accepted Date:
06 April 2022 Revised Date:
19 February 2022 Available Online:
15 February 2023
Abstract:
Finding transition metal catalysts for effective catalytic conversion of CO to CO2 has attracted much attention. MXene as a new 2D layered material of early transition metal carbides, nitrides, and carbo-nitrides is a robust support for achoring metal atoms. In this study, the electronic structure, geometries, thermodynamic stability, and catalytic activity of MXene (Mo2CS2) supported single noble metal atoms (NM = Ru, Rh, Pd, Ir, Pt and Au) have been systematically examined using first-principles calculations and ab initio molecular dynamic (AIMD) simulations. First, AIMD simulations and phonon spectra demonstrate the dynamic and thermal stabilities of Mo2CS2 monolayer. Three likely reaction pathways, Langmuir-Hinshelwood (LH), Eley-Rideal (ER), and Termolecular Eley–Rideal (TER) for CO oxidation on the Ru1- and Ir1@Mo2CS2 SACs, have been studied in detail. It is found that CO oxidation mainly proceeds via the TER mechanism under mild reaction conditions. The corresponding rate-determining steps are the dissociation of the intermediate (OCO-Ru1-OCO) and formation of OCO-Ir1-OCO intermediate. The downshift d-band center of Ru1- and Ir1@Mo2CS2 help to enhance activity and improve catalytst stability. Moreover, a microkinetic study predicts a maximum CO oxidation rate of 4.01 × 102 s-1 and 4.15 × 103 s-1 (298.15 K) following the TER pathway for the Ru1- and Ir1@Mo2CS2 catalysts, respectively. This work provides guideline for fabricating and designing highly efficient SACs with superb catalyts using MXene materials.
In recent years, CO oxidation at low temperature has aroused significant interest because of its importance in resolving the rising environmental issues instigated by CO emission from the vehicle's exhaust, inadequate combustion of fuel, and industrial processes [1-4]. Thus, CO oxidation process serves as a model for analyzing activities and stabilities of heterogeneous catalysts [5-7]. The adsorption and activation of the O2 is the key in CO oxidation reaction. Noble-metal supported catalysts, such as Pt [8-10], Pd [11-13], Rh [14, 15], Au [16, 17], Ir [18, 19], render good catalytic activity and stability for CO oxidation. Because of their high activity, selectivity and catalytic performance for a wide range of important chemical reactions, supported noble-metal catalysts on a high surface-area substrate have long been the most widely used in industry. However, the noble-metal (NM) supported catalysts are expensive, and their catalytic enactments strongly depend on the reaction environment [20, 21]. In the last few years, many researches have been devoted to synthesizing efficient single-atom catalysts (SACs) for CO oxidation at normal reaction conditions [22]. For a more cost-effective utilization of NM, the production and use of noble-metal based SACs have become a hot subject in the field of catalysis [23, 24]. Therefore, considerable efforts have been made to develop appropriate supports that can efficiently anchor single metal atoms without decreasing catalyst performance [25]. It has been demonstrated experimentally and computationally that single metal atoms can be anchored and stabilized on metal oxide supports or a suitable metal surface for catalysis [26]. As a successful approach to lower the cost and utilization of catalysts, the concept of singlet-atom catalysis was first proposed in 2011 [27], which has since then become a frontier in heterogeneous catalysis [28]. SACs anchored on various substrates have shown excellent catalytic selectivity and activity compared with nanosized metal particles [29, 30]. Even though considerable advances have been made in a few industrial reactions involving SACs, origin of superior catalytic properties, the catalytically active sites, and reaction mechanisms are still under active discussion.
SACs have produced substantial interest both experimentally and theoretically in heterogeneous catalysis. Qiao et al. described that SAC with Pt anchored on Fe2O3 surface exhibited excellent catalytic performance for CO oxidation at normal reaction conditions [27]. Many experimental and theoretical studies have shown that SAC is ideal for potential industrial applications because of well-defined active sites with high catalytic activity, stability, selectivity, and efficiency [31]. Talib et al. predicted that the SAC with Cr embedded on graphyne (GY) surface Cr1/GY exhibits superb catalytic activity for NO oxidation and reduction reaction [32]. Moses-DeBusk et al. reported theoretically and experimentally that the Pt single atom could be anchored on alumina (Al2O3) surface for CO oxidation reaction with high binding energies, good stability, and superb catalytic performance [33]. Recently, Talib et al. reported that the Fe single atom bonded with phosphotungstic acid cluster (Fe1/PTA) shows impressive catalytic activity toward ethylene epoxidation reaction at room temperature [34]. A number of computational and experimental works involve single metal atoms supported or embedded on metal oxides, pure metals, and two-dimensional (2D) materials. Currently, 2D materials have been widely studied [35-38], among which graphene [39, 40], phosphorene [41, 42], graphyne [43, 44], germanane [45-47], MoS2 [48, 49], WSe2 [50, 51], single-layer hexagonal BN [52, 53] and other transition metal dichalcogenides (TMDs) [54, 55], have been inspected as promising materials for anchoring metal atoms, which can impede the single-atoms from clustering and provide a particular surface zone for reactive centers [56].
MXenes are 2D layered materials of transition metal nitrides, carbides, or carbo-nitrides [57, 58]. In 2011, the first MXene (Ti3C2) was synthesized by etching Ti3AlC2 powders in half (50%) hydrogen fluoride (HF) solution at normal reaction conditions and had aroused much consideration because of their decent chemical and physical properties [58]. There is presently much interest in investigating whether MXenes are suitable for SACs. The bare exteriors of MXene substrates are synthetically dynamic and are frequently terminated with surface species, including O, S, F and OH species [59, 60]. The surface OH functional species can simply be converted into O terminated species and are stable at high-temperature. Therefore, MXenes could be effectively synthesized experimentally [61, 62], which have since found enormous interest in catalysis, thermoelectric insulators, energy storage devices, and topological insulators [63, 64]. Especially noteworthy is the finding that MXenes are promising support for SAC [65-69] because they are low cost, stable materials and can be easily synthesized experimentally [70, 71]. Related to this development, Zhang et al. [72] demonstrated that the SACs with Pt anchored on Mo2TiC2Tx (T represents the functional species: O, F and OH) surface exhibits excellent catalytic activity for hydrogen evolution reaction (HER). Indeed, Talib et al. recently reported that the SAC with non-noble metal Co embedded on MXene (Mo2CS2) support exhibits superb catalytic performance for CO oxidation via TER mechanism at normal reaction conditions [73].
Motivated by these results, we performed first-principles calculations to identify the active sites and explore reaction mechanisms of CO oxidation on NM@Mo2CS2 (NM = Ru, Rh, Pd, Ir, Pt, Au) SACs surface. These noble-metal are expensive despite their exceptional catalytic performance. Thus, effective use of noble-metal is critical, which makes it necessary to study SACs with high activity. It is found that Ru1- and Ir1@Mo2CS2 systems have large binding energies and moderately high diffusion barriers, which decrease the possibility for accumulation on support surface. In addition, Bader charge analysis and charge density difference are studied to assess the catalytic behavior of the NM@Mo2CS2 SACs for CO oxidation's reaction. Additional analyses reveal that Ru1- and Ir1@Mo2CS2 systems are excellent for CO oxidation because of the high diffusion barrier and bind substantially with the adsorbates (CO, O2), an essential obligation for the activation of the catalytic pathway. Assessment of reaction pathways and calculation of activation energy barriers reveal that all pathways are feasible at normal reaction temperatures. Moreover, a microkinetic study predicts a maximum CO oxidation rate of 4.01 × 102 s-1 and 4. 15 × 103 s-1 (298.15 K) following the TER pathway for the Ru1- and Ir1@Mo2CS2 SACs. Our computational study indicates that Ru1- and Ir1@Mo2CS2 are effective and promising catalysts for CO oxidation.
Computational details: The computational investigations were accomplished at the level of DFT with the Kohn-Sham spin-polarization formalism. All electronic structure calculations were performed by utilizing Vienna Ab-initio Simulation Package (VASP version 5.4) [74, 75]. The PAW (projector augmented wave) [76-78] pseudopotentials were employed to elucidate the interaction between the valence and core electrons. The van der Waals interaction was determined using the semiempirical dispersion-corrected DFT-D3 scheme by Grimme. Cut-off energy of 400 eV was set for the plane wave expansion of electronic eigenfunctions. The GGA (generalized gradient approximation) with PBE (Perdew-Burke-Ernzerhof) exchange-correlation functional was applied in these calculations [79, 80]. A cubic supercell of Mo2CS2 was used to construct the surface slab and the MXene was modelled with a supercell containing 3 × 3 primitive cell (with C = 16, Mo = 32, S = 32 atoms). A 20 Å vacuum space was used to avoid the interlayer interaction between the periodic images. The complete optimized structure of MXene with entirely relaxed positions of atoms is presented in Scheme 1.
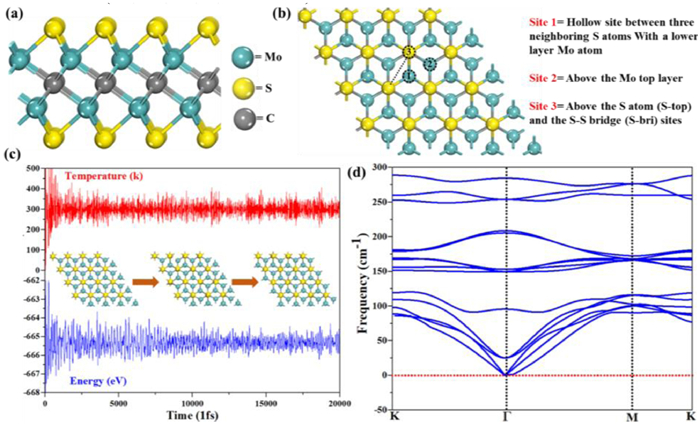
Scheme 1
Scheme 1.
The structure of Mo2CS2 monolayer: (a) Side view of the Mo2CS2 monolayer. (b) Top view and the binding sites of the Mo2CS2 (Site 1, Site 2 and Sites 3). (c) Variations of energy and temperature vs. time from AIMD simulations at 300 K for 20 ps and a time step of 2 fs. (d) Calculated phonon dispersion spectra of the Mo2CS2 monolayer.
The Γ-point was utilized for the surface Brillouin-zone integration and 5 × 5 × 1 for the partial density of state (PDOS). We have verified a k-point mesh of 3 × 3 × 1 and 5 × 5 × 1 for the surface Brillouin-zone integration (Table S2 in Supporting information) and found no significant difference (within < 0.1 eV) from the Γ-point sampling. The conjugate gradient method used the convergence of total energy and atomic forces, where the convergence criteria used were 10-5 eV and 0.02 eV/Å for total energy and atomic force, respectively. The charge density was calculated by utilizing Bader's quantum theory of atom-in-molecule (QTAIM) [81, 82].
The binding energy of noble metal (NM) atoms on MXene (Mo2CS2) is defined as:
(1)
And the adsorption energy (Eads) of an adsorbate (X) is calculated from the following equation:
(2)
where Etot[X…NM@Mo2CS2], E[NM@Mo2CS2], and E(X) represents the total energy of the X…NM@Mo2CS2 adsorption system, NM@Mo2CS2, and free adsorbate (X = CO, O2 or CO2), respectively. Furthermore, the co-adsorption energies of CO & O2, two CO2, and two O2 were evaluated as:
(3)
The transition state structures for each step were located by utilizing the climbing image nudged elastic band (CI-NEB) [83, 84], and dimer method [85, 86], with the force less than 0.02 eV/Å. The transition state structures were verified to have one imaginary vibrational frequency. The reaction energy was calculated from ∆E = EFS − EIS and the activation energy barrier (Ea) was calculated as Ea = ETS − EIS, where the EFS, EIS and ETS are the energy of the final state, initial state, and transition state, respectively. The positive/negative values represent endothermic/exothermic reactions, respectively. The ab initio molecular dynamics (AIMD) simulations were used to investigate the thermodynamic stability of the NM@Mo2CS2 surface.
Binding of NM and structures of SACs: Based on previous work [73], MXene (Mo2CS2) was chosen as a support, with noble metal (Ru, Rh, Pd, Ir, Pt and Au) atoms binding on the surface of MXene to form the SACs of NM@Mo2CS2. The completely optimized structure of the Mo2CS2 and the coordination are presented in Scheme 1. There are several adsorption sites for noble-metal atoms on Mo2CS2 as following: above the center of Mo2CS2 hexagon coordinating with the hollow site between three neighboring sulphur (S) atoms with a bottom layer Mo atom (Site-1), above the Mo top layer (Site-2), and above the sulphur top (S-top) and the S-S bridge site (Site-3). Likewise, to check the stabilities of the Mo2CS2 support, the AIMD simulations with a time step of 2 fs and phonon dispersion calculations were also performed. As presented in Scheme 1c, the Mo2CS2 support remains thermally stable at 300 K, there is no noticeable bond broken and geometric structure distortions. Moreover, the variation in the energy is very small, which confirms the thermal stability of the Mo2CS2 support.
To further ensure the lattice dynamics of the Mo2CS2 support, we perform the phonon dispersion calculations, and the corresponding results are presented in Scheme 1d. The calculated results indicate that the optical and acoustic curves are properly separated and have a positive frequency, which is consistent with the structural stability of the Mo2CS2 support. Next, we have calculated the binding energies of NM atoms for all plausible sites and found that the isolated NM atoms prefer to locate at the utmost site on the top of the S atoms (Site-1), consistent with past works [69-73]. Therefore, we focus on the catalytic activity of anchoring six noble metals (Ru, Rh, Pd, Ir, Pt and Au) atoms on Site-1 of Mo2CS2 monolayer.
To screen SACs for CO oxidation, a series of noble metal (NM1 = Ru, Rh, Pd, Ir, Pt and Au) atoms were tested. As shown in Fig. S1 (Supporting information), all the NM atoms prefer to anchor on Site-1 of the Mo2CS2 monolayer, consistent with others [70-73]. The calculated binding energies of various metal atoms anchored on Site-1 of Mo2CS2 vary from nearly −0.50 eV to −4 eV (Table S1 and Fig. S2 in Supporting information). The order of binding energies decreases as Ir > Ru > Rh > Pd > Pt > Au. That is, for the noble metals in the same period, the binding energy decrease with the increased atomic number (Ru > Rh > Pd; Ir > Pt > Au). Moreover, the cohesive energies of metal atoms were also calculated and compared to their binding energy to assess the NM stability on Mo2CS2 (Fig. S2a and Table S1). All NM binding energies are larger than their cohesive energies, implying that they prefer to be uniformly anchored on the Mo2CS2 surface rather than agglomerate. Bader charges (Fig. S2b and Table S1), bond distances (Fig. S1), electron density difference (Fig. S3 in Supporting information), and spin-polarized partial density of states (PDOS) (Fig. S4 in Supporting information) are used to assess the binding nature and stability. Ir1 and Ru1 have more strong interaction with NM@Mo2CS2 at Site-1 and form a strong bond with the adjacent S atoms with the average bond lengths of 2.20 and 2.19 Å, respectively. These noble metal atoms robustly bind to Mo2CS2, thus favoring formation of stable SACs.
We also calculated the diffusion barriers of the NM atoms on Mo2CS2 from the Site-1 to its adjacent position. The diffusion barriers were evaluated as the energy difference among the metastable configuration and the most stable one [87, 88]. The minimum energy pathways for the NM diffusion from Site-1 to its adjacent positions are displayed in Figs. S5–S10 (Supporting information). The computed diffusion energy barriers for Ru1 and Ir1 adatoms are high (1.27 and 1.26 eV), implying difficult to diffuse at room temperature, as shown in Figs. S5 and S8. From Figs. S6, S7, S9 and S10, Rh1, Pd1, Pt1 and Au1 adatoms can quickly diffuse from the Site-1 to its adjacent positions, with lower diffusion energy barriers of 0.98, 0.32, 0.58 and 0.11 eV, respectively. Therefore, Rh1, Pd1, Pt1 and Au1 adatoms would prefer to diffuse and may form clusters easily on the Mo2CS2 monolayer. Consequently, we focus on Ru1 and Ir1 atoms embedded on Mo2CS2 surface, because they are stable at room temperature, and clustering are neither kinetically nor thermodynamically preferred.
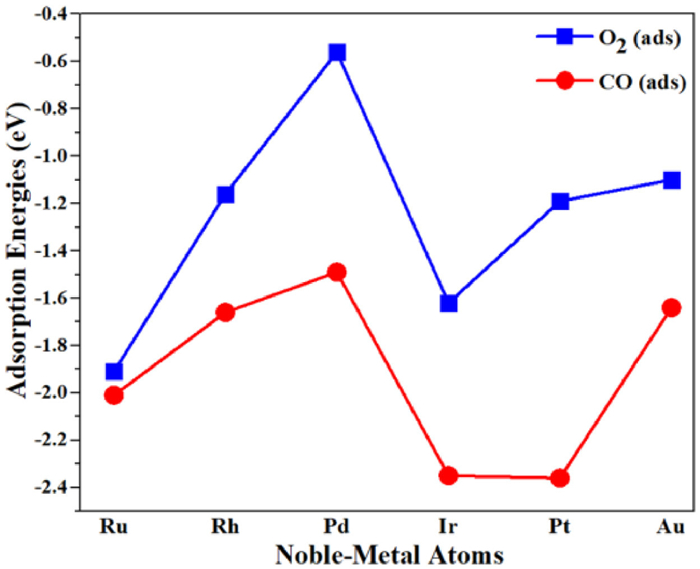
Adsorption of CO and O2: To explore CO oxidation reaction, the adsorption energies of CO and O2 on the NM@Mo2CS2 were calculated. The adsorption energies and calculated parameter of these adsorbates are presented in Fig. 1 and Table S3 (Supporting information). A detailed description of these adsorbates and structural parameters are summarized in Supporting information (see pages S11 and S12). The NM@Mo2CS2 (NM = Rh, Pd, Pt, Au) catalysts are not favorable for CO oxidation, as shown by the data depicted in Supporting information. Notably, CO and O2 adsorption on Ru1@Mo2CS2 is comparable, while all others favor the adsorption of CO instead of O2. Subsequently, all potential reaction pathways of CO oxidation on the Ru1- and Ir1@Mo2CS2 catalysts were explored.
Figure 1
Figure 1.
Adsorption energies (Ead in eV) of O2 (blue) and CO (red) on the NM@Mo2CS2 (NM = Ru, Rh, Pd, Ir, Pt and Au) surface at the Site-1.
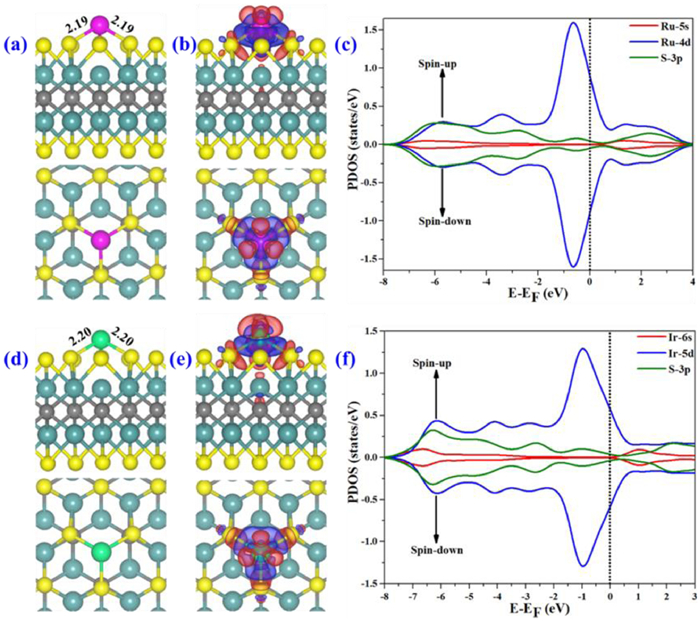
The computed geometries of the most stable Ru1- and Ir1@Mo2CS2 systems are demonstrated in Figs. 2a and d. The Ru1 and Ir1 adatom's binding at Site-1 of the Mo2CS2, with the binding energies of −3.73 eV and −3.79 eV, is comparable with the Co binding energy to Mo2CS2 support [73]. The calculated binding energies of Ru1 and Ir1 are larger than bulk's formation energies by 1.46 eV and 2.37 eV (Table S1), indicating that the single Ru1 and Ir1 atom interact strongly with the Mo2CS2 surface. The Ru1 and Ir1 atoms are attached with adjacent three S atoms with NM-S (NM = Ru1, Ir1) bond distances of 2.19 Å and 2.20 Å, respectively. The Bader charges of Ru1 and Ir1 atoms on Mo2CS2 are + 0.56 |e| and + 0.27 |e|, respectively, indicating substantial charge transfer from NM adatom's to Mo2CS2 surface as well as ionic and covalent metal-support interaction (CMSI) [89]. Meanwhile, NM adatom's positive charge stabilizes the adsorption of gases (O2, CO). The charge density difference is presented in Figs. 2b and e, which shows the charge density flows from NM towards the neighboring S atoms. The results of charge density difference are in good agreement with the Bader charge assessment. Moreover, strong interaction among NM and its nearby S atoms are confirmed by PDOS for Ru1- and Ir1@Mo2CS2, as presented in Figs. 2c and f. Clearly, the Ru1 4d orbitals overlap with S 3p orbitals, accounting for the strong covalent interaction among the Ru1 and S atoms at Site-1 of Mo2CS2. The situation of Ir1 is similar (Fig. 2f).
Figure 2
Figure 2.
(a, d) Top and side views of the optimized structure of Ru1- and Ir1@Mo2CS2. (b, e) PEDD, blue (red) isosurface shows charge addition (depletion) areas. (c, f) PDOS proposed on NM-d (blue), NM-s (red) and S-3p (green) states. The Fermi level (EF) is set at zero.
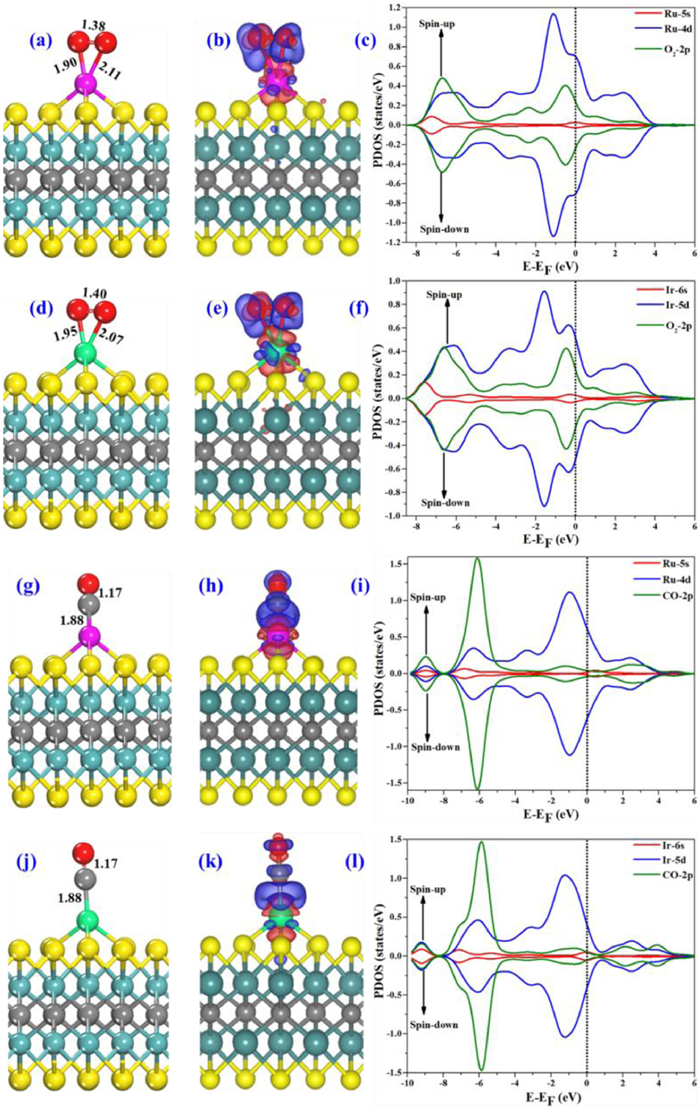
The performance of a catalyst depends on effective binding of the reactive species at the catalytic active centers. Figs. 3a and d, shows the most stable adsorption geometries of O2 on Ru1- and Ir1@Mo2CS2, where O2 has a side-on configuration. The O2 adsorption energies on the Ru1- and Ir1@Mo2CS2 surfaces were −1.91 eV and −1.62 eV, respectively, lower than CO adsorption. Here NM binds the O2 reactant more strongly than the Co1-Mo2CS2 (−1.40 eV) catalyst [73]. The average bond lengths between NM-O are ~2 Å for Ru1- and Ir1@Mo2CS2. On the other hand, the bond distance of the adsorbed O2 become larger (1.38 Å and 1.40 Å) compared with the gas phase O2 (1.23 Å), suggesting formation of superoxide (O2−). The Bader charge indicates that O2(ads) gains −0.35ǀeǀ from the surfaces of Ru1- and Ir1@Mo2CS2, and is strongly activated for further reaction. As a result, the charge transfers from NM d orbitals (4d and 5d) to antibonding 2π* orbital of O2(ads), which causes O-O bond stretch and the superoxide (O2−). The charge density difference of the O2(ads) on NM@Mo2CS2 (Figs. 3b and e) indicates charge transfer from NM@Mo2CS2 to O2, which agrees with the calculated PDOS (Figs. 3c and f). The strong mixing between NM-d orbitals and O2 2p orbital close to the EF is shown in PDOS plot.
Figure 3
Figure 3.
(a, d, g, j) Side view of the computed structure of O2 and CO on Ru1- and Ir1@Mo2CS2 through a side-on and end-on configurations. (b, e, h, k) PEDD, blue (red) isosurface shows charge addition (depletion) areas. (c, f, i, l) PDOS projected on NM-d (blue), NM-s (red) O-2p (green) and C-2p (green) states. The Fermi level (EF) is set at zero.
The adsorption of CO on NM@Mo2CS2 are also studied, where CO is adsorbed to NM dopant via end-on configuration. Figs. 3g and j demonstrate the adsorption structure of CO on NM@Mo2CS2 with adsorption energy of −2.01 and −2.35 eV. CO adsorption energies are higher than that of O2 adsorption. The calculated distances are 1.88 Å for NM-C and 1.17 Å for C–O, which is 0.03 Å larger than that of the gas phase CO (1.14 Å). Likewise, CO adsorption occurs through donation and back-donation bonding model [90, 91]. With this model, while the lone pair of CO donates to the empty d/s orbitals of NM atom, the filled NM d orbitals interact with the low-lying 2π* antibonding orbitals of CO via back donation. According to the Bader charges, the adsorbed CO gets −0.38 |e| and −0.36 |e| from the Ru1- and Ir1@Mo2CS2, which is in line with the charge density difference (Figs. 3h and k): increase between NM and C while decrease between NM and S. Figs. 3i and l indicate that NM and CO interaction is mainly due to the strong overlap of the NM d orbitals with the low-lying 2π* orbitals of the adsorbed CO orbitals near the EF. The strong mixing and charge transfer between the NM and CO lead to the C–O bond activation.
Fig. S17a (Supporting information) elucidates the highly stable co-adsorption configuration of CO+O2, two CO's, and two O2 molecules on Ru1- and Ir1@Mo2CS2, where O2 adsorbs with the end-on configuration. The binding energies of co-adsorption are −2.99 and −2.97 eV for CO + O2 and −3.93 and −4.23 eV for two CO's, respectively, on Ru1- and Ir1@Mo2CS2. The co-adsorption energies of two CO's are higher than the co-adsorbed CO+O2 and the individual O2 (−1.91 and −1.62 eV) and CO (−2.01 and −2.35 eV), indicating that the co-adsorption is highly favored. Furthermore, the co-adsorption of two CO's is better than that of CO+O2. We noted that the co-adsorption energy of two O2 molecules is−2.10 eV (Ru1) and−1.68 eV (Ir1), respectively, lower than the co-adsorption energy of two CO and CO + O2, suggesting that CO can easily co-adsorb with another O2 rather than the two O2. The charge density difference of CO + O2, two CO's and two O2's on NM@Mo2CS2 are presented in Fig. S17d (Supporting information). The calculations indicate that CO co-adsorption through C atoms on NM@Mo2CS2 is the most desirable configuration (Fig. S17a).
Reaction mechanism of CO and O2: With these results, we have explored the likely reaction mechanisms for CO oxidation. In the ER pathway, the O2 first adsorbs on NM@Mo2CS2 surface, while CO from the gas phase directly interacts with the pre-adsorbed and activated O2 to form CO2. In the LH pathway, the reactant molecules O2 and CO co-adsorb at the adjacent positions of NM@Mo2CS2 surface, initiating peroxide-like (OCOO) intermediate, which dissociates into CO2 as a final product. Consequently, the adsorption energy of CO is somewhat larger than that of O2 on NM@Mo2CS2, and the co-adsorption energy of CO and O2's is greater than the individual CO and O2 adsorption. In the TER pathway, the two reactant CO molecules first co-adsorbed on the NM@Mo2CS2 surfaces and the third free O2 interacts directly from the gas phase with pre-adsorbed and activated species forming an OCO-NM-OCO pentagonal like intermediate, which dissociates into two CO2's. The co-adsorption energy of two CO's (−3.93 eV and −4.23 eV) are larger than the co-adsorption energy of O2 + CO (−2.9 eV and −2.97 eV) on Ru1- and Ir1@Mo2CS2, indicating that the TER pathway is the most promising for CO oxidation on the NM@Mo2CS2 surfaces.
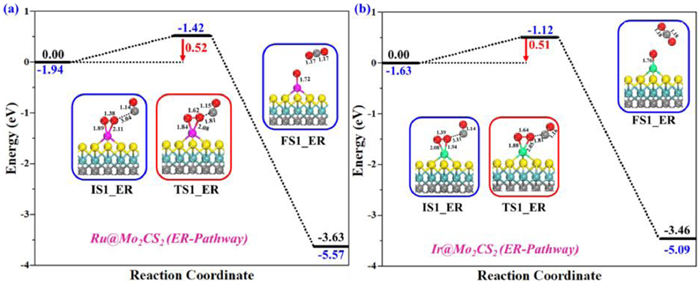
Although CO has better interaction with NM@Mo2CS2 than O2, under oxygen-rich environment ER mechanism cannot be excluded for CO oxidation process. We therefore have also investigated the ER pathway's feasibility for CO oxidation as an initial step. The computed structures about the ER pathway, comprising the initial state (IS1-ER), transition state (TS1-ER), final state (FS1-ER), and the potential energy profile are shown in Figs. 4a and b. The first step is the adsorption of O2 on NM@Mo2CS2, as described earlier (Figs. 3a and d). With CO reacting via the ER mechanism, "CO3-like" intermediate or CO2 will be formed and in the second step NM-oxo complex ([NM]O). This mechanism is possible thermodynamically as the adsorption of O2 on NM@Mo2CS2 surface are highly exothermic [−3.63 eV (Ru1), −3.46 eV (Ir1)]. CO attacking the pre-adsorbed O2 on the NM@Mo2CS2 surface (Figs. 4a and b) is chosen as an initial state (IS1-ER), where CO is 3.04 Å and 3.11 Å apart from one O atom of O2 molecule. The O2 accepts a side-on configuration and is activated substantially for CO molecule. In the catalytic cycle, the IS1-ER corresponds to CO physically adsorbed on a chemically adsorbed O2 molecule, which then interacts with O2 to produce [NM]-O-O…CO (TS1-ER). The side-on O2 is changed to end-on configuration during the formation of FS1-ER via TSI-ER with reaction energy barriers of 0.52 and 0.51 eV (IS1-ER → FS1-ER), respectively. The imaginary frequency of TS1-ER is at 570i cm-1 and 541i cm-1, which is related to formation of the C–O bond and the cleavage of O–O bond. In the TS1-ER, the O–O bond length is further stretched from 1.38 Å, and 1.39 Å (superoxide) to 1.62 Å and 1.64 Å (peroxide), and a new C–O bond starts to progress. The FS1-ER depicts the oxygen transfer from O2(ads) to CO on NM@Mo2CS2 surface and the progress of a new C–O bond (1.81 Å). At last, the O–O bond gets cleaved, producing a physically adsorbed CO2 and an atomic Oa that remains on the NM@Mo2CS2 surface. This step (IS1-ER → FS1-ER) is highly exothermic with ∆E = −3.63 eV and −3.46 eV, respectively. The CO2 in FS1-ER shows weak interaction with the residing Oa atom, and hence it could be desorbed efficiently from NM@Mo2CS2 surface, forming Oa covered free surface for another CO oxidation.
Figure 4
Figure 4.
The reaction energy profiles for CO oxidation to yield first CO2 on (a) Ru1- and (b) Ir1@Mo2CS2 surfaces via ER pathway. All energies are specified in eV. Side views of the optimized configurations of initial structure (IS1), transition state (TS1), and final state (FS1) of the ER pathway. All bond lengths are in Å.
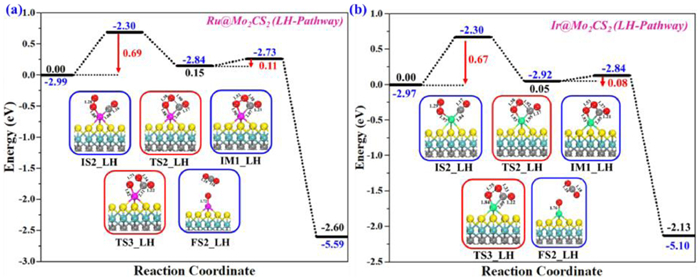
Figs. 5a and b illustrate the potential energy profiles as well as the computed geometries of the corresponding stationary points of initial state (IS2-LH), transition states (TSs-LH), intermediate (IM-LH), and final state (FS2-LH) for CO oxidation via LH mechanism. The most stable CO and O2 co-adsorption configuration was chosen as an initial state (IS2-LH), where both O2 and CO are adsorbed at the NM@Mo2CS2 surface. In IS2-LH structure, NM is connected to one of the oxygen atoms (Oa) of O2 and C atom of CO with distances of 1.88 and 1.88 Å (Ru1), 1.97 and 1.84 Å (Ir1), respectively (Figs. 5a and b). The C–O bond length changes slightly to 1.16 Å and 1.17 Å, whereas the O–O bond length in IS2-LH is stretched to 1.28 Å and 1.29 Å (superoxide). In the catalytic cycle, the free end of the activated O2 starts to interact with the C atom of CO to generate OOCO (peroxide-like) intermediate (IM1-LH) via transition state (TS2-LH), and the O–O bond stretched from 1.28 and 1.29 Å to 1.52 and 1.53 Å (peroxide) with the formation of a new C–O bond (1.38 Å and 1.37 Å) between O2 and CO molecules. The computed activation energy barriers (imaginary frequencies) and reaction energies for the IS2-LH→IM1-LH conversion are 0.69 (384i cm-1) and 0.67 eV (371i cm-1), and 0.15 and 0.05 eV (endothermic), respectively, for Ru1 and Ir1. The first step is the rate-determining step of the LH pathway. Next, the O–O bond dissociation of IM1-LH intermediate (OOCO) happens instantly, then CO2 desorbs and the remaining atomic Oa adsorbed on NM@Mo2CS2 surface. The calculated activation energy barriers for the IM1-LH→FS2-LH conversion is merely 0.11 (348i cm-1) and 0.08 eV (309i cm-1), and the exothermic reaction energies of −2.60 and −2.13 eV, respectively. All the energy barriers for the TSs (TS2-LH and TS3-LH) are appropriate for CO oxidation under ambient reaction conditions.
Figure 5
Figure 5.
The reaction energy profiles for CO oxidation to yield CO2 on (a) Ru1- and (b) Ir1@Mo2CS2 surfaces via LH pathway. All energies are specified in eV. Side views of the optimized structures of initial state (IS2), transition states (TS2 & TS3), intermediate (IM1), and final state (FS2) of the LH pathway. All bond lengths are in Å.
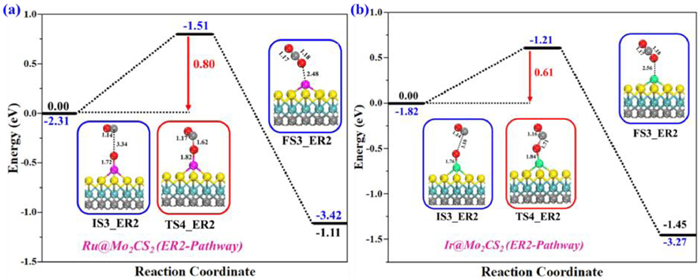
With the first CO2 desorption, the Oa atom on NM@Mo2CS2 surface with the NM-O bond distance of 1.72 and 1.76 Å, respectively. This Oa oxidizes the second CO via the second ER pathway (denoted as ER2 pathway hereafter). The potential energy surfaces and computed structures of resultant stationary points such as initial structure (IS3-ER2), transition state (TS4-ER2), and final structure (FS3-ER2) for second CO oxidation are depicted in Figs. 6a and b. A parallel configuration of the physiosorbed CO resides 3.34 and 3.10 Å away from the Oa atom pre-adsorbed on NM@Mo2CS2 is chosen as an initial structure (IS3-ER2). The Oa atom is strapped away from the NM atom when it reacts with CO, resulting in a second CO2 (FS3-ER2) via TS4-ER2. The calculated activation energy barriers and reaction energies for the IS3-ER2 → FS3-ER2 conversion are 0.80 (580i cm-1) and 0.61 (554i cm-1) eV, and −1.11 and −1.45 eV (exothermic), respectively for Ru1 and Ir1. In addition, the formed CO2 is weakly adsorbed with 2.48 and 2.56 Å apart from the NM@Mo2CS2 surface and easily desorbed and the catalyst is recovered for a new CO oxidation cycle.
Figure 6
Figure 6.
The reaction energy profiles for CO oxidation to yield the second CO2 on (a) Ru1- and (b) Ir1@Mo2CS2 surfaces via ER2 pathway. All energies are specified in eV. Side views of the optimized structures of the initial state (IS3), transition state (TS4), and final state (FS3) of the ER2 mechanism. All bond lengths are in Å.
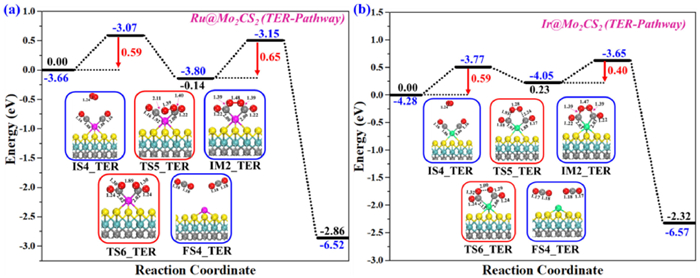
The TER pathway as a new pathway for CO oxidation with SACs was firstly reported by Mao et al. [92, 93] on single Au1 adatom anchored by h-BN surface, in which two co-adsorbed CO's react with a free O2 on Au1@h-BN to generate a pentagonal-like OCO−Au1−OCO intermediate that dissociates into two CO2's. The potential energy curves of the TER pathway and the computed geometries of the initial state (IS4-TER), transition states (TSs-TER), intermediate (IM2-TER), and final state (FS4-TER) are presented in Figs. 7a and b. As the co-adsorption energy of two CO molecules on NM@Mo2CS2 is much higher than that of CO + O2, the TER pathway is dominant for CO oxidation on the NM@Mo2CS2 surface. The most stable adsorption configuration of the O2 lying atop the two pre-adsorbed CO's on NM@Mo2CS2 surface was chosen as an initial structure (IS4-TER). In IS4-TER, the average bond distance of C…O (physically adsorbed O2 and chemically pre-adsorbed CO) is 3.16 and 3.26 Å, and the O−O distance extended from 1.23 (free O2) to 1.24 Å. In the catalytic cycle, the O–O distance is extended from 1.24 to 1.48 (Ru1) and 1.47 Å (Ir1) (superoxide) when O2 simultaneously approaches two C atoms of the chemisorbed CO's. As shown in Figs. 7a and b, the bond distance between O2 and C gradually declines, and two new C–O bonds are formed, leading to a pentagonal ring structure (OCO–NM–OCO) intermediate (IM2-TER) through TS5-TER. The corresponding activation energy barrier (TS5-TER) and reaction energy for the IS4-TER→IM2-TER are calculated to be 0.59 (372i cm−1) and 0.59 (334i cm−1) eV, and −0.14 (exothermic) and 0.23 eV (endothermic). Finally, the OCO–NM–OCO intermediate dissociates to generate two physically adsorbed CO2's by cleavage of O–O bond, through the transition state TS6-TER. The subsequent energy barrier (TS6-TER) and exothermic reaction energy for the IM2-TER→FS4-TER conversion are calculated to be 0.65 (430i cm−1) and 0.40 (429i cm−1) eV, and −2.86 and −2.32 eV, respectively, for Ru1 and Ir1 systems. Interestingly, the final step is the rate-determining step of the Ru1@Mo2CS2 TER reaction pathway, whereas in the case of Ir1@Mo2CS2 the first step is the rate-determining step. Hence, it can be concluded that all energy barriers and reaction energies are reasonable for CO oxidation. The comparatively low activation barrier indicates that CO oxidation may occur via the TER pathway at low-temperature. Additionally, the adsorption energy of the generated CO2 is only −0.17 eV, suggesting that CO2 can desorb easily into the air at low temperature and the NM@Mo2CS2 surface is ready for a new CO oxidation catalytic cycle.
Figure 7
Figure 7.
The reaction energy profiles for CO oxidation yields two CO2 molecules on (a) Ru1@Mo2CS2 and (b) Ir1@Mo2CS2 surfaces via the TEH pathway. All energies are specified in eV. Side views of the optimized configuration of initial structure (IS4), transition states (TS5 and TS6), intermediate (IM2), and final state (FS4) of the TER mechanism. All bond lengths are in Å.
To further understand the superb catalytic reactivity of O2 with the pre-adsorbed CO molecules on NM@Mo2CS2, we carried out electronic structure analysis via partial density of states (PDOS). Figs. S18a–c and S19a–c (Supporting information) show PDOS of the initial configuration (IS4), intermediate (IM2) and transition state (TS5) of TER pathway. As shown in Figs. S18c and 19c, the antibonding 2π* orbital of O2 is partially occupied, which activates O2 and significant weakens O–O bond. Furthermore, compared with the initial configuration (IS4), O2 2p orbitals in transition state (TS5) strongly mix with the C 2p orbitals over the entire energy region, which confirm the conclusion that the O2 can be activated by the pre-adsorbed CO's. From the frontier molecular orbital analysis as shown in Figs. S18d and 19d (Supporting information), the LUMO (lowest unoccupied molecular orbital) of the physically adsorbed O2 matches with the HOMO (the highest occupied molecular orbital) of the chemosorbed two CO's on the NM@Mo2CS2, which facilitates electron transfer and activating O–O double bond. The PDOS of the d-band center (ɛd) of the Ru1 and Ir1 atoms were calculated as presented in Figs. S20, S21 and Table S4 (Supporting information). The d-band center can define the interaction strength among the adsorbates and NM@Mo2CS2 catalyst. The valence electrons of NM atoms play an important role in forming and breaking bonds among the single-atom catalyst and adsorbates. Figs. S20 and S21 show that the higher interaction between NM@Mo2CS2 and adsorbate, the d-band shifts to the lower energy with respect to the Fermi level. The calculated results indicate that the pre-adsorbed two CO molecules initiate the physically adsorbed O2 and the d-band center moves to the lower energy.
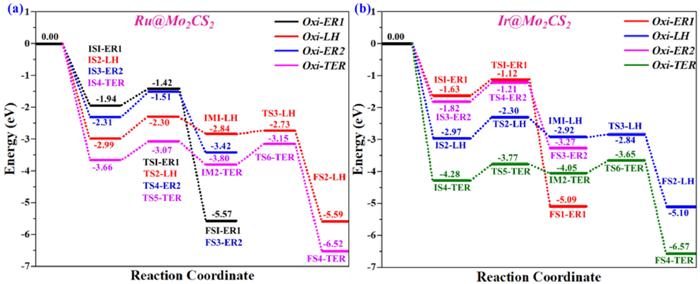
In summary, the ER, LH, and TER mechanisms all have relatively low activation energy (Ea) barriers in the range of 0.50–0.80 eV (ER), 0.08–0.70 eV (LH), and 0.40–0.65 eV (TER), respectively. The schematic energy profiles for complete reaction pathways for CO oxidation on the NM@Mo2CS2 surfaces are précised in Fig. 8. The results show that CO oxidation is feasible on NM@Mo2CS2 SAC under ambient reaction conditions through the LH [0.69 and 0.11 eV (Ru1), 0.67 and 0.08 eV (Ir1)], ER (0.52 and 0.80 eV (Ru1), 0.51 and 0.61 eV (Ir1)] and TER [0.59 and 0.65 eV (Ru1), 0.59 and 0.40 eV (Ir1)] pathways, respectively. Due to the higher activity of two co-adsorbed CO molecules on the NM@Mo2CS2 surface, CO oxidation via TER mechanism is dominant and favorable both thermodynamically and kinetically. The NM@Mo2CS2 supported SACs are theoretically predicted to be highly reactive catalyst for CO oxidation reaction. This results also agrees with findings reported elsewhere on MXene supported SACs for various reactions [94-97].
Figure 8
Figure 8.
Schematic energy profiles compared to the structures accomplished in Fig. 5, Fig. 6, Fig. 7 along the minimum energy surface for CO oxidation on (a) Ru1@Mo2CS2 and (b) Ir1@Mo2CS2 surfaces. All energies are given regarding the reference energy, i.e., the sum of energies of the NM@Mo2CS2, one O2 and two gaseous CO's.
Reaction rate and kinetics: To gain more comprehensive knowledge of the catalytic CO oxidation reaction on NM@Mo2CS2 system, the rate constants k at different temperatures for all the elementary steps involved in LH, ER, and TER pathways are calculated through the transition state (TS) theory by using the following equation:
(4)
In the above equation, kB is the Boltzmann constant (kB = 1.3806452 × 10-23 J/K), T is the absolute temperature (T = 298.15 K), h is the Planck's constant (h = 6.626 × 10−34 J s), Ea is the activation energy, and qR & qTS are the vibrational partition functions for reactants of the elementary steps and for the transition state, respectively. The vibrational partition function q was calculated by using the following equation:
(5)
In the above equation, the v denotes the frequency of vibrational mode i, with the imaginary frequency being excluded for TS. The rate constants of all the elementary steps involved in three reaction pathways (LH, ER and TER) under various temperatures are presented in Table 1. It is found that the rate constants of the rate-determining step in the TER pathway are much higher than that of the LH and ER pathways. That is, the TER pathway proceeds much easier as compared to the other pathways, in excellent agreement with the computed energy barriers from DFT calculations. However, the rate constants for various reactions increase at high temperatures, implying that NM@Mo2CS2 can accelerate CO oxidation at relatively higher temperatures. On the other hand, the rate constant for all elementary steps involved in three reaction pathways abruptly decreases at 100 K, indicating that the cryogenic condition inhibits CO oxidation with O2.
Table 1
Table 1.
Rate constant k (s−1) of all the elementary steps involved in three reaction pathways: LH, ER and TER on NM@Mo2CS2 surfaces under various temperatures.
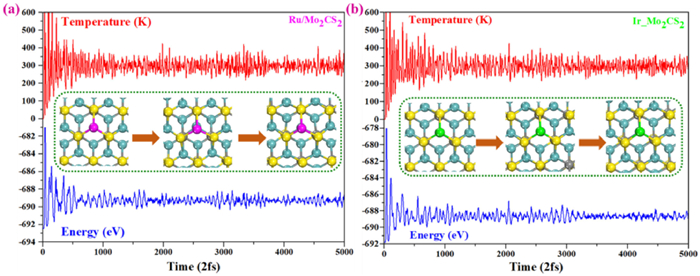
The thermodynamic stability of NM@Mo2CS2 catalysts are further calculated by using AIMD simulations with a time step of 2 fs. As presented in Fig. 9 and Fig. S22 (Supporting information), the Ru1- and Ir1@Mo2CS2 systems remain thermally stable at 300 K; there is no noticeable geometric structure change. On the other hand, the Rh1-, Pd1-, Pt1-, and Au1@Mo2CS2 systems show the geometric structure distortion, thermodynamically unstable at 300 K. Therefore, we conclude that the designed Ru1- and Ir1@MXene based catalysts are stable and hold potential for experimental design. These results are also in good agreement with the above-mentioned diffusion barriers calculations.
Figure 9
Figure 9.
Variations of energy and temperature versus time, for AIMD simulations at 300 K for 20 ps and a time step of 2 fs: (a) Ru1@Mo2CS2, (b) Ir1@Mo2CS2. The dashed line box presents top views of the initial, intermediate, and final atomic configurations.
In conclusion, the catalytic performance of MXene (Mo2CS2) supported SACs for noble-metal atoms (Ru, Rh, Pd, Ir, Pt and Au) has been investigated by using first-principles calculations. Through AIMD simulations and phonon dispersion spectra, the thermal and dynamical stabilities of the Mo2CS2 support are verified. Our results indicate that Ru1 and Ir1 are better than other noble metals (Rh1, Pd1, Pt1 and Au1) in terms of possessing considerable binding energies with Mo2CS2 support and moderately high diffusion barriers, making them stable SACs. Adsorption energies and Bader charge analysis of O2 and CO adsorbed on the NM@Mo2CS2 were discussed. CO oxidation on Ru1- and Ir1@Mo2CS2 SACs via three well-known pathways (ER, LH, TER) are investigated in detail. In the ER pathway, the physiosorbed CO interacts with the remaining Oa atom on NM@Mo2CS2 to produce CO2 with the activation barriers of 0.80 and 0.61 eV as the rate-determining step. In the LH pathway, the co-adsorbed CO(ad) + O2(ad) to form CO2 with the activation barriers of 0.69 and 0.67 eV as the rate-determining step. A pentagonal ring like intermediate OCO–NM–OCO forms a physiosorbed O2 on two chemisorbed CO's in the TER pathway. The pentagonal ring like intermediate dissociates with an activation barrier of 0.65 eV as the rate-determining step. It is found that TER (0.65, and 0.59 eV) pathway is thermodynamically more favorable than LH (0.69, and 0.67 eV) and ER (0.80, and 0.61 eV) pathways with a lower activation barrier.
Ab initio molecular dynamics simulations and microkinetic analysis for CO oxidation on NM@Mo2CS2 SACs also show higher stability and catalytic feasibility at for CO oxidation. The NM@Mo2CS2 (NM = Ru1, Ir1) SACs are theoretically predicted to be highly effective for CO oxidation. These theoretical results provide valuable guideline for experimentalists to design noble metal single-atom catalysts for CO oxidation reaction. With these results, one can speculate that further investigations of single-cluster catalysts (SCC) [98-100] with atomically precise metal clusters firmly anchored on MXene may also be fruitful for catalyzing complicated chemical reactions.
Declaration of competing interest
The authors declare no conflicts of interests.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (Nos. 11874141 and 22033005), the Henan Overseas Expertise Introduction Center for Discipline Innovation (No. CXJD2019005), and the Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002). The calculations were performed on resources provided by the High-Performance Computing Center of Henan Normal University and using supercomputers at Tsinghua National Laboratory for Information Science and Technology. The authors are grateful to funding support from the Researchers Supporting Project number (No. RSP-2021/399), King Saud University, Riyadh, Saudi Arabia.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.04.010.
J. Liu, H. Xiao, X. Zhao, et al., CCS Chem. (2022), 10.31635/ccschem. 022.202201796. doi: 10.31635/ccschem.022.202201796
Scheme 1
The structure of Mo2CS2 monolayer: (a) Side view of the Mo2CS2 monolayer. (b) Top view and the binding sites of the Mo2CS2 (Site 1, Site 2 and Sites 3). (c) Variations of energy and temperature vs. time from AIMD simulations at 300 K for 20 ps and a time step of 2 fs. (d) Calculated phonon dispersion spectra of the Mo2CS2 monolayer.
Figure 2
(a, d) Top and side views of the optimized structure of Ru1- and Ir1@Mo2CS2. (b, e) PEDD, blue (red) isosurface shows charge addition (depletion) areas. (c, f) PDOS proposed on NM-d (blue), NM-s (red) and S-3p (green) states. The Fermi level (EF) is set at zero.
Figure 3
(a, d, g, j) Side view of the computed structure of O2 and CO on Ru1- and Ir1@Mo2CS2 through a side-on and end-on configurations. (b, e, h, k) PEDD, blue (red) isosurface shows charge addition (depletion) areas. (c, f, i, l) PDOS projected on NM-d (blue), NM-s (red) O-2p (green) and C-2p (green) states. The Fermi level (EF) is set at zero.
Figure 4
The reaction energy profiles for CO oxidation to yield first CO2 on (a) Ru1- and (b) Ir1@Mo2CS2 surfaces via ER pathway. All energies are specified in eV. Side views of the optimized configurations of initial structure (IS1), transition state (TS1), and final state (FS1) of the ER pathway. All bond lengths are in Å.
Figure 5
The reaction energy profiles for CO oxidation to yield CO2 on (a) Ru1- and (b) Ir1@Mo2CS2 surfaces via LH pathway. All energies are specified in eV. Side views of the optimized structures of initial state (IS2), transition states (TS2 & TS3), intermediate (IM1), and final state (FS2) of the LH pathway. All bond lengths are in Å.
Figure 6
The reaction energy profiles for CO oxidation to yield the second CO2 on (a) Ru1- and (b) Ir1@Mo2CS2 surfaces via ER2 pathway. All energies are specified in eV. Side views of the optimized structures of the initial state (IS3), transition state (TS4), and final state (FS3) of the ER2 mechanism. All bond lengths are in Å.
Figure 7
The reaction energy profiles for CO oxidation yields two CO2 molecules on (a) Ru1@Mo2CS2 and (b) Ir1@Mo2CS2 surfaces via the TEH pathway. All energies are specified in eV. Side views of the optimized configuration of initial structure (IS4), transition states (TS5 and TS6), intermediate (IM2), and final state (FS4) of the TER mechanism. All bond lengths are in Å.
Figure 8
Schematic energy profiles compared to the structures accomplished in Fig. 5, Fig. 6, Fig. 7 along the minimum energy surface for CO oxidation on (a) Ru1@Mo2CS2 and (b) Ir1@Mo2CS2 surfaces. All energies are given regarding the reference energy, i.e., the sum of energies of the NM@Mo2CS2, one O2 and two gaseous CO's.
Figure 9
Variations of energy and temperature versus time, for AIMD simulations at 300 K for 20 ps and a time step of 2 fs: (a) Ru1@Mo2CS2, (b) Ir1@Mo2CS2. The dashed line box presents top views of the initial, intermediate, and final atomic configurations.
Table 1.
Rate constant k (s−1) of all the elementary steps involved in three reaction pathways: LH, ER and TER on NM@Mo2CS2 surfaces under various temperatures.

 DownLoad:
DownLoad:


 下载:
下载:











