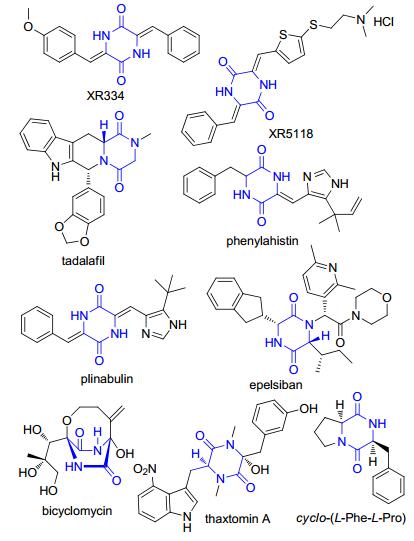
图 1.
具多种生物活性的二酮哌嗪类化合物
Figure 1.
Chemical structures of DKPs with diverse bioactivities

二酮哌嗪类化合物(diketopiperazines, DKPs)的特征结构是由两个α-氨基酸通过肽键缩合而成的最小环肽, 即环二肽(cyclic dipeptides or cyclodipeptides, CDPs).肽类天然产物是成药化合物重要的来源之一[1, 2].与其线性类似物相比, DKPs由于具有稳定的六元环结构, 良好的蛋白酶抗性和构象刚性, 表现出抗细菌[4, 5]、抗真菌[6, 7]、抗病毒[8]、抗肿瘤[9, 10]、免疫抑制[11]、神经保护[12]、抗疟疾[13]、抗朊病毒[14]、抗高血糖[15], 以及作为潜在的血脑屏障跨膜载体[16]等多种显著的生物活性及药理活性[3, 18].如nocazine家族XR334结构修饰后得到的XR5118可逆转纤溶酶原激活物抑制剂-1 (plasminogen activator inhibitor-1, PAI-1)的抑制作用, 表现出较好的溶血栓作用, 有望成为第四代溶栓药物[22, 16];具有显著抗革兰氏阴性菌活性的双环霉素(bicyclomycin), 是目前已知的唯一天然产物来源的转录终止因子Rho蛋白的选择性抑制剂[55];在天然产物phenylahistin结构基础上, 经构效关系研究合成的海洋抗肿瘤候选药物普那布林(plinabulin), 已进入临床三期研究[17, 18];分离自感染疮痂病土豆切片的thaxtomin A目前已被美国环保署登记为一种生物除草剂[19].目前已进入临床使用的DKPs类药物有:用于治疗肺动脉高压和勃起功能障碍的它达拉非(tadalafil)[20]以及催产素受体拮抗剂epelsiban等[21] (图 1).同时, 基于CDPs的小分子量及较强的跨膜运输能力, 已有研究人员提出CDPs可作为微生物种内及种间群体感应的化学信号, 参与自然界中的生物化学通讯现象.如霍乱弧菌(Vibrio cholera)产生的致病因子主要有两种:霍乱毒素(cholera toxin, CT)和毒素协同菌毛(toxin-coregulated pilus, TCP), 而这两种致病因子的表达都受ToxR操纵子的调控, 由创伤弧菌(Vibrio vulnificus)产生的cyclo-(L-Phe-L-Pro)能够下调ToxR操纵子以抑制CT和TCP的产生[23].
合成可作为药物先导物的二酮哌嗪类化合物已日益引起人们的关注.近年来国内外研究人员在DKPs有机合成方面开展了大量的研究工作, 建立了组合化学、固相合成和液相合成等一系列方法与技术, 成功获得了具有多种药理活性的DKPs[1, 24].天然产物的生物合成研究是拓宽其化学结构多样性的一个极为有效的途径[25].与其有机合成相比, DKPs的生物合成研究起步较晚, 但随着现代生物技术和高通量测序技术的飞速发展, 人们对DKPs生物合成分子机制与酶学机理的认识不断深入, 若干DKPs类化合物的生物合成途径也已被阐明, 通常可分为非酶催化和酶催化途径.
非酶催化途径是指肽链的环化是自发的, 未经酶的参与.如普遍存在于高等哺乳动物中枢神经系统的cyclo-(L-His-L-Pro)[59].酶催化途径可分为非核糖体肽合成酶(non-ribosomal peptide synthetases, NRPSs)和环二肽合酶(cyclodipeptide synthases, CDPSs)生物合成途径两大类[27, 28].目前多数报道的DKPs是通过NRPS途径合成或是NRPS途径中的副产物.典型的NRPSs是一类多酶复合物, 由不同模块组成, 每个模块负责将一个氨基酸缩合到肽链上.一个基本的延伸模块至少含有3个功能结构域[29]:负责底物识别和活化的腺苷化结构域(adenylation domain, A domain), 通过硫酯键加载活化底物的肽基载体蛋白结构域(peptide carrier protein domain, PCP domain), 以及负责肽键缩合的缩合结构域(condensation domain, C domain).此外, 某些模块还包含其它修饰功能结构域, 如甲基化结构域(N-methy- lation domain, M domain)和异构化结构域(epimerization domain, E domain), 最后通常由负责肽链解离的硫酯酶结构域(thioesterase domain, TE domain)将聚肽链从PCP上解离下来.相比之下, CDPS途径是新发现的一种与NRPS途径不同的DKPs类化合物生物合成途径. Gondry和Sylvie课题组[27]于2002年在诺尔斯氏链霉菌(Streptomyces noursei)中克隆了白诺氏菌素(albonoursin)的生物合成基因簇, 发现其中albC基因编码的大小仅为239 aa的酶能够催化albonoursin骨架结构cyclo- (L-Phe-L-Leu)的合成.这个小分子蛋白与NRPSs几乎没有同源性, 并与任何已知功能蛋白均无相似性[39]. 2009年, AlbC首次在体外被鉴定为环二肽合酶, 成为第一个功能被阐明的CDPS[32].与NRPSs不同的是, CDPSs本身不具备活化氨基酸的催化活性, 而是直接以细胞内氨酰-tRNAs (aa-tRNAs)作为底物, 竞争蛋白质生物合成所需的aa-tRNAs合成次级代谢产物, 将初级代谢和次级代谢直接关联.本文对近年来国内外有关环二肽合酶(CDPSs)生物合成途径研究进展进行了综述.
CDPSs通常是由200~300个氨基酸残基组成的小分子蛋白, 大部分CDPSs序列同源性都很低, 氨基酸一致性小于30%.截至2017年6月, 通过序列同源性检索鉴定出约800个CDPSs编码基因, 且其数量在稳步增加.它们分布在所有域(domain)中, 但主要集中在3个细菌门(Actinobacteria, Firmicutes和Proteobacteria).这其中, 约400个CDPSs来源于放线菌门[34].
CDPSs分布在系统发育树的两个主要分支上(图 2).依据特定催化残基不同分为两大亚家族: NYH亚家族(含“N40, Y202, H203”)和XYP亚家族(含“X40, Y202, P203”).序列比对显示Y202残基在所有活性CDPSs中都严格保守.在NYH亚家族中, 97%的CDPSs的N40是保守的, H203在95%的CDPSs中保守.在XYP亚家族的CDPSs中, X40可以是Q (38.5%), N (25%), K (13.5%), S (15.4%)或A (7.7%)[41a], P203几乎是严格保守的, 只有来源于Parabacteroides的CDPS(WP_ 005863245.1)在相应位置为Q[33].随着鉴定的CDPSs数量的不断增加, 对其亚家族的分类也在不断调整与完善.
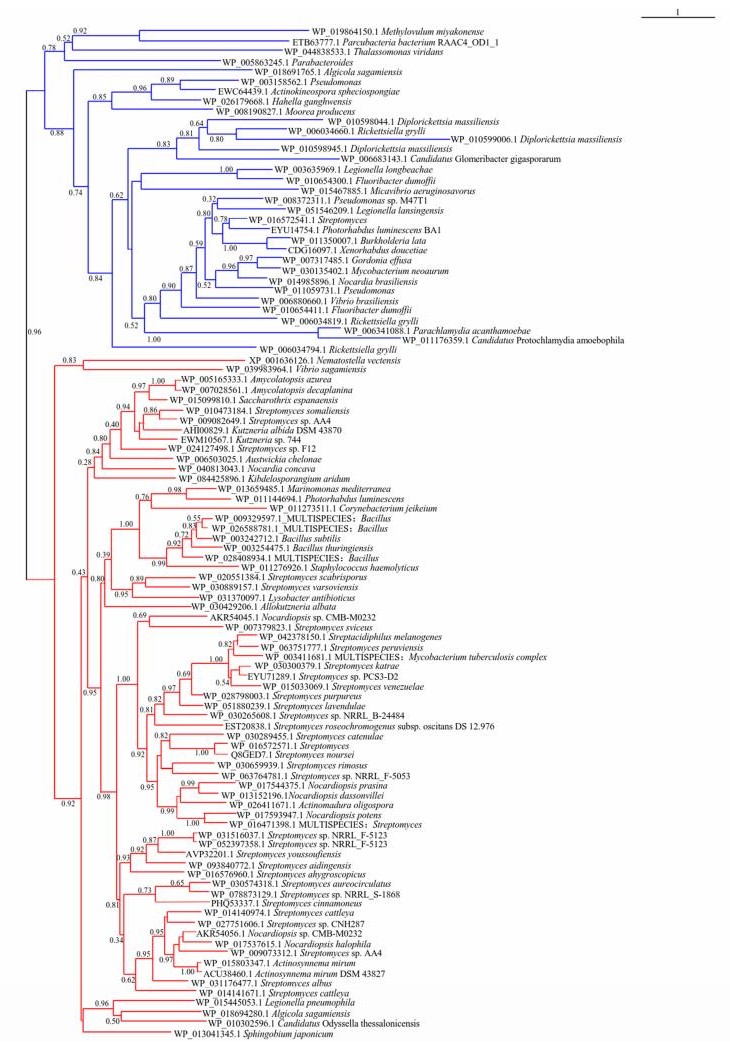
The tree was generated using the PhyML program (v3.1) based on the maximum likelihood method. CDPSs' names consist of the corresponding accession numbers in the NCBI database and the host organisms in which they were found. The two main branches corresponding to the XYP and NYH subfamilies are shown in blue and red, respectively
最早解析的三个CDPSs的晶体结构均属NYH亚家族, 分别是来自S. noursei的AlbC (PDB ID, 3OQV)[30], Mycobacterium tuberculosis的Rv2275 (PDB ID, 2X9Q)[36a]以及Bacillus licheniformis的YvmC-Blic (PDB ID, 3OQH)[49](图 3A). AlbC和YvmC为单体结构, Rv2275为二聚体结构.尽管这些CDPSs序列一致性低于27%, 但它们的晶体结构能较好地重叠, rms偏差仅为2.27 Å (Rv2275 vs. AlbC, over 192 Cα atoms), 2.2 Å (Rv2275 vs. YvmC, over 211 Cα atoms)以及2.46 Å (AlbC vs. YvmC, over 180 Cα atoms).每个CDPS单体重叠性较好的结构为5条β折叠(β3-β7)及2个α螺旋(α2和α4), 其中β3-β5, α2和α4组成Rossmann折叠结构域[35d](图 3A).三个CDPSs都具有一个相似的表面结合口袋(surface-accessible pocket) P1, 且口袋内保守的催化残基也重叠性较好(除Y202外). CDPSs的结构与class-I aa-tRNA合成酶(aa-tRSs)相似, 尤其类似于class-Ic TyrRSs和TrpRSs[36](图 3B), 均具有Rossmann折叠结构域, CDPSs的P1口袋与二者的氨酰基结合口袋位置相对应[46]. CDPSs和I型aaRSs之间也存在若干关键差异: CDPSs不具有I型aa-tRSs中tRNA-结合结构域, 其依靠表面的氨基酸残基结合tRNA; TyrRSs和TrpRSs一般形成同源二聚体, 两个活性位点位于二聚体交界面, 而CDPSs单体即可有活性.因此, CDPSs可能由I型aaRSs进化而来, 从而能以aa-tRSs的产物为底物催化酰胺键形成.近期Bourgeois等[41a]解析了XYP亚家族的3个CDPSs的晶体结构, 分别为来自Nocardia brasiliensis的Nbra- CDPS, Rickettsiella grylli的Rgry-CDPS及Fluoribacter dumoffii的Fdum-CDPS.两个亚家族的CDPSs晶体结构差异性集中体现在Rossmann折叠结构域的前半部分(α1, β2, β6, β7, α8), 其中α8的空间位置在两个亚家族CDPSs中都不保守, 而且它的长度在XYP- CDPSs中更长, 二者Rossmann折叠结构域的后半部分(α2, α4-α7, β3-β5)则重叠性较好(图 3C).关键催化残基(S37, Y202, Y178和E182)的空间位置在两个亚家族是相同的, 这说明两个亚家族的催化机制可能是一致的[41a].
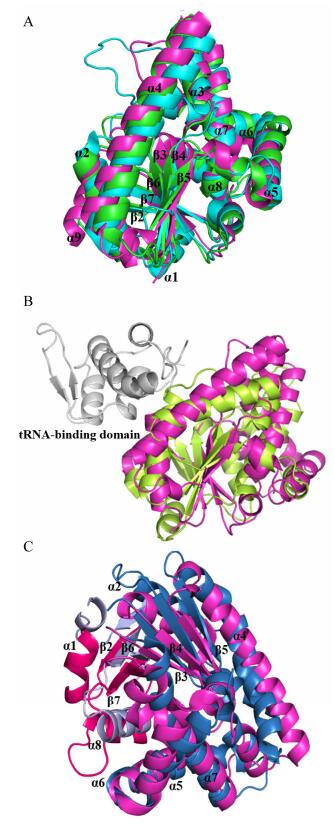
(A) Superimposition of the cartoon structures of AlbC (PDB ID, 3OQV; light magenta), Rv2275 (PDB ID, 2X9Q; green), and YvmC (PDB ID, 3OQH; cyan). (B) Superimposition of the cartoon structures of AlbC and TyrRS of M. jannaschii (PDB ID, 1J1U). AlbC is in light magenta; TyrRS is in lemon; a C-terminal domain involved in tRNA-binding and anticodon recognition coloured in gray. (C) Superimposition of the cartoon structures of AlbC and Rgry-CDPS (PDB ID, 5MLP). The first half of the Rossmann fold of AlbC is in hot pink and Rgry-CDPS is in light blue; the second half of the Rossmann fold of AlbC is in light magenta and Rgry-CDPS is in sky blue
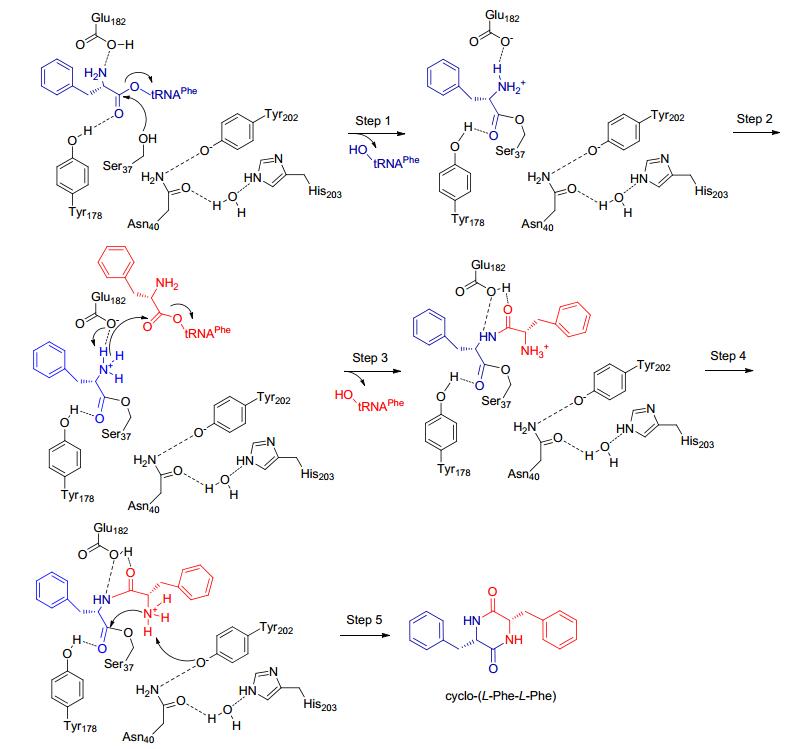
AlbC的相关结构研究揭示了CDPSs可能的催化机制为:以Phe-tRNAPhe和Leu-tRNALeu/Phe-tRNAPhe为底物, 通过一个连续的乒乓反应(Ping-pang reaction)合成环二肽[37c](Scheme 1).以合成cyclo-(L-Phe-L-Phe)为例.首先, 第一个Phe-tRNAPhe进入AlbC的P1口袋, tRNA上带负电的核糖磷酸与AlbC的α4螺旋上带正电的氨基酸残基相互作用[37], 其苯丙氨酰基部分与E182和Y178侧链羟基之间的氢键作用使其固定于P1中, 随后S37的羟基亲核攻击Phe-tRNAPhe的酯羰基, 氨酰基部分转移到保守的S37残基上形成苯丙氨酰-AlbC中间体.之后, 由CL1 (catalytic loop 1: residues 36-43), CL2 (catalytic loop 2: residues 202-207), α6 C末端(residues 152~ 159)和α7中部(residues: 175~183)形成一个可容纳第二个aa-tRNA的P2口袋.第二个Phe-tRNAPhe的tRNA结构部分与AlbC的α6-α7相互作用, 其氨酰基部分进入P2口袋, E182将苯丙氨酰-AlbC中间体的氨基氮去质子化, 使其可以攻击第二个Phe-tRNAPhe的酯羰基, 形成二肽基-AlbC中间体.最后, Y202去质子化二肽基-AlbC中间体的氨基氮, 进行分子内环化, 最终形成cFF[26, 56].此过程中, N40和H203与水分子形成氢键网络, 对P2口袋的稳定和催化残基的精确定位具有重要作用[35].
目前由18种天然氨基酸(除Asp及Lys之外)与tRNAs结合形成的氨酰-tRNAs作为底物产生的75种环二肽中, 多数含有疏水性氨基酸, 未检测到酸性Asp及碱性Lys残基, 最近的报道中有检测到Arg残基[31].研究人员发现大多数CDPSs似乎首先特异性识别aa- tRNAs的氨酰基[37], 与此一致, AlbC负责合成cyclo- (L-Phe-L-Xaa), 载有D-Phe的tRNAs不被AlbC转化, 这表明只有L型氨基酸可作为AlbC的底物[46].同时, AlbC优先识别疏水性氨基酸, 而不能识别带电或极性的氨基酸. 2014年, Moutiez等[26]通过对携带野生型及突变型大肠杆菌序列的aa-tRNAs进行生化实验, 研究了AlbC与两个底物间的相互作用.实验结果表明, AlbC识别第一个底物的特异性由其氨酰基部分决定, 识别第二个底物的特异性涉及氨酰基部分和tRNAs的氨基酸接受茎序列, 特别是N1-N73碱基对. tRNAs部分可用于激活氨基酸的羧基, 促进氨酰基-酶中间体的形成, 并通过静电相互作用增强底物与CDPSs的结合. CDPSs底物结合口袋中的氨基酸残基对识别底物具有非常重要的作用. 2011年, Sauguet等[30]的实验结果显示AlbC的P1口袋内L200突变为Gln导致产物由cyclo-(L-Phe-L-Leu)转变为cyclo-(L-Tyr-L-Leu), 这直接证明了P1口袋内L200对识别第一个底物发挥关键作用.最近, Yao等[51]发现了首个以cWV为主要产物的来自于Streptomyces youssoufiensis OUC6819的CDPS DmtB1, 能够合成cyclo-(L- Trp-L-Xaa) (Xaa=Val, Pro, Leu, Ile和Ala), 系列定点突变结果显示位于P1口袋的L185突变为Phe后, 产物比例均发生明显变化, 主产物变为cWP, 即影响了对第二个底物的识别, 位于P2口袋的V205被Met取代后, cWL成为体内合成的主产物.体外实验结果也证实了L185及V205对第二个底物识别的重要作用.
随着鉴定的CDPSs序列数量日益增加, 如何预测CDPSs底物特异性, 合理推测其产物的问题摆在了研究人员面前.两个底物结合位点的揭示为第一种预测模型的发展奠定了基础[32].该预测模型的建立是基于已鉴定功能的CDPSs中构成P1和P2口袋的残基的多序列比对结果(表 1).其中P1口袋含8个残基(33-35-65-67- 119-185-186-200, AlbC numbering), P2口袋含7个残基(152-155-156-159-204-206-207, AlbC numbering).模型将CDPSs分为六大类:主产物分别为cyclo-(L-Leu- L-Leu) (cLL), cyclo-(L-Trp-L-Trp) (cWW), cyclo-(L-Cys- L-Cys) (cCC), cyclo-(L-Ala-L-Glu) (cAE)的CDPSs以及主产物中只确定了一个氨基酸cyclo-(L-X-L-Glu) (cXE)和cyclo-(L-Ala-L-X) (cAX)的CDPSs.自从这项研究以来, 推定CDPSs的数量增加了50%以上[33~45], 同时, 模型也进一步得到了扩充, 增加了主产物为cyclo-(L- Tyr-L-Tyr) (cYY), cyclo-(L-Leu-L-X) (cFX), cyclo-(L-Trp- L-X) (cWX)和cyclo-(L-Cys-L-X) (cCX)等的分类[31].然而, 研究人员的实验结果显示许多CDPSs的预测产物与其实际产物相差甚远, 如在Li等[50]的研究中, 模型预测显示WP_078950527.1、WP_019889609.1和WP_063768158.1这三个CDPSs的产物为cFX, 而其实际产物均为cWW.该预测模型需通过鉴定更多的CDPSs的结构及结合口袋中的氨基酸残基, 同时需考虑tRNA与底物间相互作用来得到进一步的完善.
 下载:
导出CSV
下载:
导出CSV
| Specificity group (*) | Consensus motif for P1 | Consensus motif for P2 |
| cLL (NYH) (7/123) |  |
 |
| cWW (NYH) (8/22) |  |
 |
| cCC (NYH) (8/60) |  |
 |
| cAE (XYP) (8 / 55) |  |
 |
| cXE (XYP) (9/81) |  |
|
| cAX (XYP) (10/88) |  |
|
| a The numbers in brackets (*) correspond to the number of CDPSs experimentally characterized vs. the total number of CDPSs in the group (Last up-date: June 2017, Gondry et al. 2018)[40]; degenerated positions are indicated in gray. | ||
不同于NRPSs识别底物的多样性, 且某些NRPSs自身含有修饰氨基酸的结构域, CDPS生物合成途径的修饰发生在环二肽合成之后, 由CDPS生物合成基因簇中的后修饰酶基因对环二肽进行修饰[39].
目前国内外已研究的经CDPS途径合成的二酮哌嗪类化合物共报道了6例[34].包括albonoursin[40a, 27, 32]、mycocyclosin[29, 32, 40]、pulcherriminic acid[41]、nocazine家族nocazine XR334和nocazine E [60]、bicyclomycin[54]以及drimentines[51].此外, 甲基化双色氨酸及nocardioazines A和B的合成途径也已有所研究[38, 44].
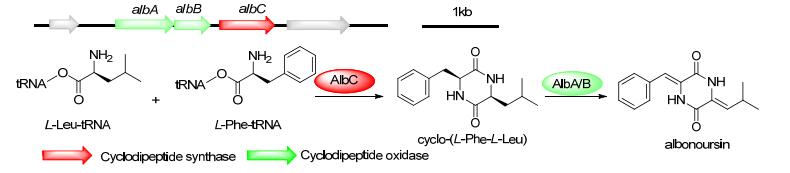
来源于Streptomyces noursei的albonoursin生物合成基因簇包含3个基因:编码CDPS的albC及编码环二肽氧化酶(cyclic dipeptide oxidase, CDO)的albA和albB. AlbC催化Phe-tRNAPhe和Leu-tRNALeu缩合形成cyclo- (L-Phe-L-Leu), 随后通过AlbA/AlbB氧化产生albonoursin(图 5)[27]. CDO是黄素依赖性α, β-脱氢酶, 具有保守的FMN结合位点, 底物特异性低, 可催化albonoursin中两个双键的形成[40a].单独的albA表达并不能产生具活性的CDO, 其必须要求albB的共同参与. AlbB可能通过作为电子受体或催化AlbA的翻译后修饰发挥作用, 其确切的反应机理尚未阐明[27, 32].
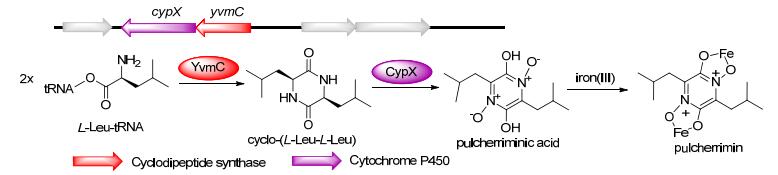
来源于Bacillus subtilis的pulcherriminic acid的生物合成基因簇包含一个编码CDPS的yvmC基因和一个编码细胞色素P450酶的基因cyp134A1 (cypX). YvmC以两分子的Leu-tRNALeu为底物合成cyclo-(L-Leu-L-Leu), 细胞色素Cyp134A1(CypX)催化发生三步氧化形成双N-氧化物, 即发生羟基化及连续两次脱水或直接的电子转移反应, 同时伴随着DKP环的芳构化(图 6)[41]. Pulcherriminic acid能够通过其两个异羟肟酸部分螯合Fe3+, 推测其可以作为抗生素或铁载体[41c].
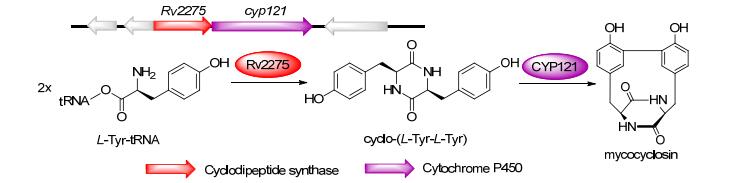
来源于M. tuberculosis的mycocyclosin生物合成基因簇包含2个基因:编码CDPS的rv2275与编码细胞色素P450酶的rv2276 (cyp121). Rv2275以两分子的Tyr-tRNATyr为底物合成cyclo-(L-Tyr-L-Tyr), CYP121在体外反应中催化环二肽酪氨酸侧链的两个β-C之间的C—C单键形成, 产生mycocyclosin[36a, 40b](图 7). Mycocyclosin的结构中两个酪氨酸侧链位于DKP杂环的同一侧, 这在酶催化过程中需要通过旋转Cα—Cβ键而实现. CYP121与一系列唑类抗真菌药物(例如克霉唑, 咪康唑)紧密结合, 表明它可能可以作为M. tuberculosis等结核病原体中抗生素的新靶点[48].
来源于Nocardiopsis dassonvillei和Nocardiopsis alba的nocazine XR334和nocazine E生物合成基因簇包含4个基因:编码CDPS的Ndas_1148、编码CDO同源基因的Ndas_1146/Ndas_1147及编码O-甲基转移酶(methyltransferase, MT)的Ndas_1149. Giessen等[42, 60]通过体外实验证明了Ndas_1148催化合成cyclo-(L- Phe-L-Tyr), Ndas_1146/Ndas_1147催化一次或两次α, β-脱氢, Ndas_1149进一步催化酪氨酸羟甲基化形成nocazine XR334和nocazine E(图 8).该基因簇中CDO的上游还存在一个可能的O-甲基转移酶基因Ndas_1145.
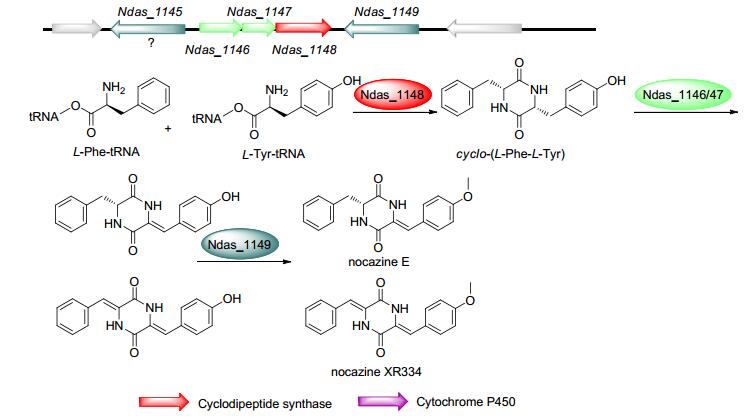
The function of gene Ndas_1145 remains to be clarified
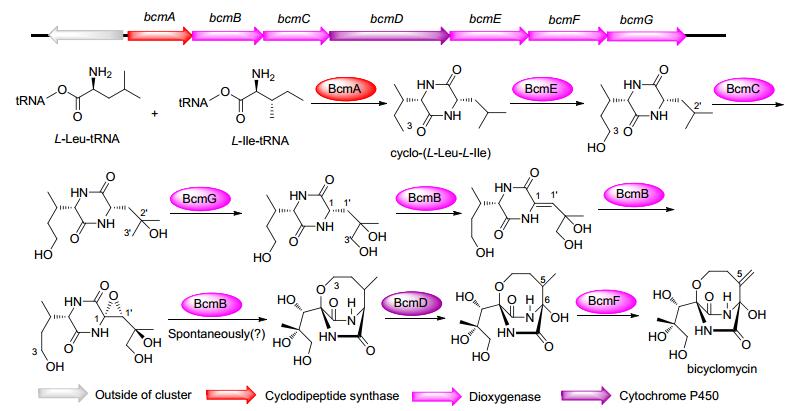
双环霉素bicyclomycin含有高度氧化的双环, 是一种广谱抗生素.其生物合成基因簇广泛分布在放线菌门和变形菌门, 甚至也存在于近百株条件致病菌绿脓杆菌(Pseudomonas aeruginosa)的基因组中[58].其生物合成途径中共包含7个酶: 1个环二肽合酶BcmA, 6个氧化还原酶(包括5个α-酮戊二酸/ Fe2+依赖性双加氧酶BcmB, BcmC, BcmE, BcmF和BcmG, 以及1个细胞色素P450单加氧酶BcmD) (图 9).首先, BcmA以Leu-tRNALeu和Ile-tRNAIle为底物催化合成cyclo-(L-Leu-L-Ile); 随后, 分别经BcmE, BcmC和BcmG催化C-3, C-2', C-3'位上羟基化.接着BcmB催化C1-C1'脱氢形成双键环氧化, 进一步氧杂桥环的形成是否由BcmB催化或自发进行还未确定. BcmD负责在桥联的sp3-杂化的碳中心上加载叔羟基, BcmF催化产生环外双键最终形成双环霉素[54].
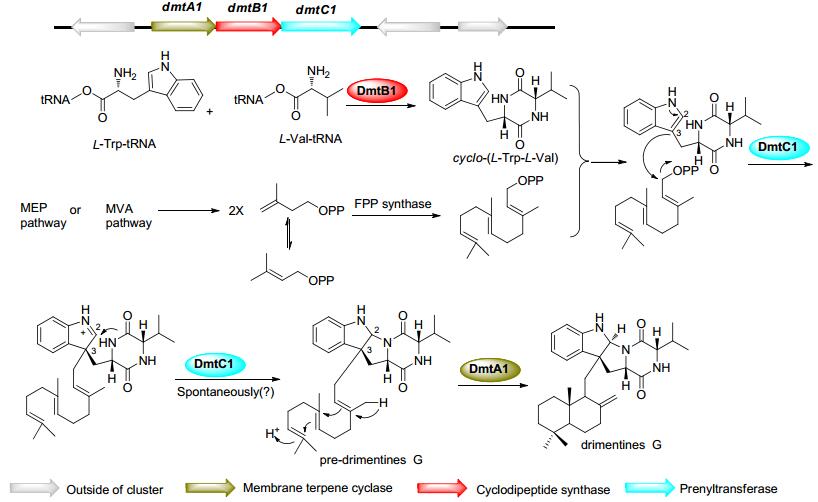
本课题组采用异源表达、基因阻断、互补和高表达以及体外生化实验等手段对3个同源的含有CDPS的基因簇dmt1-3进行了研究, 揭示了一类杂合异戊烯化DKPs类化合物drimentines的生物合成途径.以drimentine G为例, 环二肽合酶DmtB1首先催化产生cyclo-(L-Trp-L-Val), 接着PSL (phytoene-synthase-like)家族异戊烯转移酶DmtC1将法尼基焦磷酸(farnesyl pyrophosphate, FPP)加载到cyclo-(L-Trp-L-Val)的C-3位上, 形成pre-drimentine G, 然后经膜蛋白萜烯环化酶DmtA1催化双键起始的环化反应形成drimentine G(图 10)[51].
此外, 其他两个CDPS生物合成途径也已有所研究, 它们的环二肽产物特征为cyclo-(L-Trp-L-Xaa). Actinosynnema mirum基因组中含有一个编码CDPS基因Amir_4627、一个编码N-甲基转移酶基因Amir_4628以及一个编码脂肪酰基-CoA连接酶基因Amir_4626组成的基因簇[38, 43]. Amir_4627催化产生cyclo-(L-Trp-L-Trp), 它被甲基转移酶Amir_4628修饰产生带有一个或两个N-甲基化的环二肽.在体内及体外实验中, 脂肪酰基- CoA连接酶Amir_4626均被验证没有发挥功能[38].来源于Nocardiopsis sp. CMB-M0232的nocardioazines A/B生物合成所需的基因分布在两个基因簇内, 包含编码CDPS的NozA、可能参与后修饰的异戊烯转移酶NozC、甲基转移酶NozB及细胞色素P450酶NozD及NozE, 编码后修饰酶的基因具体功能还未阐明[34, 44].
对CDPS生物合成途径的产物进行结构多样性的合理设计, 以期获得结构新颖或良好生物学活性的药物先导物已成为近年来CDPS领域研究的热点之一.目前主要有两种方法可合理修饰肽天然产物结构.
第一种方法为改变CDPS催化产生DKPs骨架的多样性.由于CDPSs的特异性与tRNAs结合的氨酰基部分及其tRNAs序列有关, 因此可以通过改变tRNAs携带的氨基酸序列或改变结合氨基酸的tRNAs序列来实现.后者可通过体外转录或采用突变技术将特定序列变化引入tRNAs中, 在不影响其完整结构的同时产生序列多样性的tRNAs.前者则可改变组成氨基酸的种类、数量或连接性.由于最初发现的CDPSs只能催化天然aa- tRNAs, 因此, 研究人员使用非常见氨基酸(non-canonical Amino Acids, ncAAs)代替培养基中的天然氨基酸, 结合使用营养缺陷型表达宿主, 产生非天然CDPs[45].截至目前, 已发现26个ncAAs可作为CDPSs的底物, 从而产生约200个非天然环二肽[25].
第二种方法为在DKP骨架上通过修饰酶引入不同的修饰[47].在CDPS途径开展组合生物合成时, 可优先考虑CDPS生物合成基因簇中的修饰酶. CDPS基因簇中已经鉴定出氧化还原酶(AlbA/B, CypX)[27, 41b]、甲基转移酶(Ndas_1149, Amir_4628)[42, 38]、异戊烯基转移酶(DmtC1)[51]等修饰酶, 多表现出相对宽泛的底物选择性. Skinnider等[43]对739个CDPS基因簇进行分析, 发现CDPS周围还存在大量N-乙酰基转移酶、糖基转移酶等修饰酶基因, 这表明将来乙酰化、糖基化的DKPs化合物会被不断发现, 为进一步采用组合生物合成和合成生物学策略以增加天然产物多样性提供了重要功能元件.
二酮哌嗪类化合物以其丰富的生物活性引起了人们极大的关注, 自2002年首次发现后, tRNA依赖性环二肽合酶(CDPSs)生物合成途径已在国内外研究领域取得了显著进展.但已阐明的CDPS生物合成途径的数量远远低于CDPSs的多样性.随着CDPSs编码基因不断发现及生物合成途径的逐步揭示, 更多新颖的后修饰酶将被挖掘和表征.通过组合生物合成等研究进行结构多样性的合理设计, 获得结构丰富的具生物活性的DKPs类化合物将尤为重要.在此过程中, 由于异戊烯化的天然产物通常具有明显不同于其非异戊烯化前体的生物活性, 这使得异戊烯转移酶对于构建具有生物活性的“非天然”天然产物的合成途径有很大潜力[51~53, 57].同时, 关于CDPS的一些其他问题有待于解答, 包括是否有特异性因素决定CDPS会产生一系列CDPs还是只有一种化合物[2]?通过P1、P2关键残基预测环二肽产物的模型, 未将tRNA与底物相互作用考虑进来, 还不能普遍应用, 能否形成更加完善的预测方法?对P1、P2口袋选择底物的特异性这一问题的认知还在不断更新及发展[31].未来亚家族的分类是否还会有所扩充及调整, 仍不得而知.但毋庸置疑的是, 这些棘手待解决问题将迅速推动着CDPS生物合成途径的研究前行, 为最终实现人为创造和生产高活性DKPs化合物提供重要的理论依据.
Borthwick, A. D. Chem. Rev. 2012, 112, 3641. doi: 10.1021/cr200398y
Giessen, T. W.; Marahiel, M. A. Int. J. Mol. Sci. 2014, 15, 14610. doi: 10.3390/ijms150814610
Menegatti, S.; Hussain, M.; Naik, A. D. Biotechnol. Bioeng. 2013, 110, 857. doi: 10.1002/bit.24760
de Carvalho, M. P.; Abraham, W. R. Curr. Med. Chem. 2012, 19, 3564. doi: 10.2174/092986712801323243
Kohna, H.; Widger, W. Curr. Drug Targets: Infect. Disord. 2005, 5, 273. doi: 10.2174/1568005054880136
Musetti, R.; Polizzotto, R.; Vecchione, A. Micron 2007, 38, 643. doi: 10.1016/j.micron.2006.09.001
Ström, K.; Sjögren, J.; Broberg, A. Appl. Environ. Microbiol. 2002, 68, 4322. doi: 10.1128/AEM.68.9.4322-4327.2002
Rodriguez, P. L.; Carrasco, L. J. Virol. 1992, 66, 1971.
Kanoh, K.; Kohno, S.; Katada, J. Bull. Agric. Chem. Soc. Jpn. 1999, 63, 1130.
Kanzaki, H.; Yanagisawa, S.; Nitoda, T. Bull. Agric. Chem. Soc. Jpn. 2004, 68, 2341.
Waring, P.; Beaver, J. Gen. Pharmacol. 1996, 27, 1311. doi: 10.1016/S0306-3623(96)00083-3
Cornacchia, C.; Cacciatore, I.; Baldassarre, L. Mini-Rev. Med. Chem. 2012, 12, 2. doi: 10.2174/138955712798868959
Pérezpicaso, L.; Olivo, H. F.; Argotteramos, R. Bioorg. Med. Chem. Lett. 2012, 22, 7048. doi: 10.1016/j.bmcl.2012.09.094
Bolognesi, M. L.; Ai, T. H.; Staderini, M. ChemMedChem 2010, 5, 1324. doi: 10.1002/cmdc.201000133
Song, M. K.; Hwang, I. K.; Rosenthal, M. J. Exp. Biol. Med. 2003, 228, 1338. doi: 10.1177/153537020322801112
王贵鑫, 血栓与止血学, 2011, 17, 234. doi: 10.3969/j.issn.1009-6213.2011.05.014Wang, G. X. Chin. J. Thromb. Hemostasis. 2011, 17, 234 (in Chinese). doi: 10.3969/j.issn.1009-6213.2011.05.014
孙天文, 丁忠鹏, 王世潇, 中国海洋药物, 2016, 4, 79.Sun, T. W.; Ding, Z. P.; Wang, S. X. Chin. J. Mar. Drug. 2016, 4, 79 (in Chinese).
Martins, M. B.; Carvalho, I. Tetrahedron 2007, 63, 9923. doi: 10.1016/j.tet.2007.04.105
Zhang, H.; Ning, X.; Hang, H. Org. Lett. 2013, 15, 5670. doi: 10.1021/ol4026556
Wen, L.; Liu, Q. Q.; Yang, Q. H. Prog. Mod. Biomed. 2012, 119, 2894.
Rajesh Shinghal, M. D.; Allison Barnes, M. S.; Mahar, K. M. J. Sex. Med. 2013, 10, 2506. doi: 10.1111/jsm.12272
Charlton, P. A.; Faint, R. W.; Bent, F. Thromb. Haemostasis. 1996, 75, 808. doi: 10.1055/s-0038-1650371
Park, D. K.; Lee, K. E.; Baek, C. H. J. Bacteriol. 2006, 188, 2214. doi: 10.1128/JB.188.6.2214-2221.2006
(a) Wyatt, P. G.; Allen, M. J.; Borthwick, A. D. Bioorg. Med. Chem. Lett. 2005, 15, 2579.
(b) Fischer, P. M. J. Pept. Sci. 2003, 9, 9.
(c) O'Neill, J. C.; Blackwell, H. E. Comb. Chem. High Throughput Screening. 2007, 10, 857.
(d) Merwe, E. V. D.; Huang, D.; Peterson, D.; Kilian, G.; Milne, P. J.; Venter, M. V. D. Peptides 2008, 29, 1305.
Canu, N.; Belin, P.; Thai, R. Angew. Chem., Int. Ed. 2018, 57, 3118. doi: 10.1002/anie.201712536
Moutiez, M.; Seguin, J.; Fonvielle, M.; Belin, P.; Jacques, I. B.; Favry, E.; Arthur, M.; Gondry, M. Nucleic Acids Res. 2014, 42, 7247. doi: 10.1093/nar/gku348
Lautru, S.; Gondry, M.; Genet, R. Chem. Biol. 2002, 9, 1355. doi: 10.1016/S1074-5521(02)00285-5
Moutiez, M.; Belin, P.; Gondry, M. Chem. Rev. 2017, 117, 5578. doi: 10.1021/acs.chemrev.6b00523
李文利, 夏娟, 微生物学通报, 2014, 41, 111.Li, W. L.; Xia, J. Microb. China 2014, 41, 111 (in Chinese).
Sauguet, L.; Moutiez, M.; Li, Y.; Belin, P.; Seguin, J.; Le Du, M.H.; Thai, R.; Masson, C.; Fonvielle, M.; Pernodet, J. L. Nucleic Acids Res. 2011, 39, 4475. doi: 10.1093/nar/gkr027
Gondry, M.; Jacques, I. B.; Thai, R. Front. Microb. 2018, 9.
Gondry, M.; Sauguet, L.; Belin, P. Nat. Chem. Biol. 2009, 5, 414. doi: 10.1038/nchembio.175
Jacques, I. B.; Moutiez, M.; Witwinowski, J. Nat. Chem. Biol. 2015, 11, 721. doi: 10.1038/nchembio.1868
(a) Brockmeyer, K.; Li, S. M. J. Nat. Prod. 2017, 80, 2917.
(b) Mukai, T.; Reynolds, N. M.; Crnković, A. Life 2017, 7, 8.
(c) Alqahtani, N.; Porwal, S. K.; James, E. D. Org. Biomol. Chem. 2015, 13, 7177.
(d) Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J. Mol. Syst. Biol. 2011, 7, 539.
(a) Bonnefond, L.; Arai, T.; Sakaguchi, Y. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3912.
(b) Yaremchuk, A.; Kriklivyi, I.; Tukalo, M. EMBO J. 2002, 21, 3829.
(c) Jacques, I.; Seguin, J.; Moutiez, M. New Biotechnol. 2014, 31, S74.
(d) Cusack, S.; Berthet-Colominas, C.; Härtlein, M.; Nassar, N.; Leberman, R. Nature 1990, 347, 249.
(a) Vetting, M. W.; Hegde, S. S.; Blanchard, J. S. Nat. Chem. Biol. 2010, 6, 797.
(b) Han, W.; Gao, J. F. Chem. Bioeng. 2013, 30, 17 (in Chinese).
(韩伟, 高菊芳, 化学与生物工程, 2013, 30, 17.
(a) Seguin, J.; Moutiez, M.; Li, Y. Chem. Biol. 2011, 18, 1362.
(b) Minelli, A.; Bellezza, I.; Grottelli, S.; Galli, F. Amino Acids 2008, 35, 283.
(c) Moutiez, M.; Schmitt, E.; Seguin, J. Nat. Commun. 2014, 5, 5141.
Giessen, T. W.; von Tesmar, A. M.; Marahiel, M. A. Biochemistry 2013, 52, 4274. doi: 10.1021/bi4004827
(a) Aravind, L.; De, S. R. F.; Iyer, L. M. Biol. Direct 2010, 5, 48.
(b) Belin, P.; Moutiez, M.; Lautru, S. Nat. Prod. Rep. 2012, 29, 961.
(c) Roback, P.; Beard, J.; Baumann, D. Nucleic Acids Res. 2007, 35, 5085.
(a) Gondry, M.; Lautru, S.; Fusai, G. Eur. J. Biochem. 2001, 268, 1712.
(b) Belin, P.; Le Du, M.H.; Fielding, A.; Lequin, O.; Jacquet, M.; Charbonnier, J. B.; Lecoq, A.; Thai, R.; Courcon, M.; Masson, C. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7426.
(c) Leys, D.; Mowat, C. G.; Mclean, K. J. J. Biol. Chem. 2003, 278, 5141.
(d) Seward, H. E.; Roujeinikova, A.; Mclean, K. J. J. Biol. Chem. 2006, 281, 39437.
(a) Bourgeois, G.; Seguin, J.; Babin, M. J. Struct. Biol. 2018.
(b) Cryle, M. J.; Bell, S. G.; Schlichting, I. Biochemistry 2010, 49, 7282.
(c) Tang, M. R.; Sternberg, D.; Behr, R. K. Ind. Biotechnol. 2006, 2, 66.
Zhang, Q.; Li, S.; Chen, Y.; Tian, X.; Zhang, H.; Zhang, G.; Zhu, Y.; Zhang, S.; Zhang, W.; Zhang, C. J. Antibiot. 2013, 66, 31. doi: 10.1038/ja.2012.88
Skinnider, M. A.; Johnston, C. W.; Merwin, N. J. BMC Genomics. 2018, 19, 45. doi: 10.1186/s12864-018-4435-1
James, E. D.; Knuckley, B.; Alqahtani, N. ACS Synth. Biol. 2016, 5, 547. doi: 10.1021/acssynbio.5b00120
Johnson, J. A.; Lu, Y. Y.; Van Devente, J. A. Curr. Opin. Chem. Biol. 2010, 14, 774. doi: 10.1016/j.cbpa.2010.09.013
Giessen, T. W.; Altegoer, F.; Nebel, A. J.; Steinbach, R. M.; Bange, G.; Marahiel, M. A. Angew. Chem., Int. Ed. 2015, 54, 2492. doi: 10.1002/anie.201410047
Giessen, T. W.; Marahiel, M. A. Front. Microb. 2015, 6, 785.
McLean, K. J.; Cheesman, M.R.; Rivers, S.L.; Richmond, A.; Leys, D.; Chapman, S. K.; Reid, G. A.; Price, N. C.; Kelly, S. M.; Clarkson, J.; Smith, W. E.; Munro, A. W. J. Inorg. Biochem. 2002, 91, 527. doi: 10.1016/S0162-0134(02)00479-8
Bonnefond, L.; Arai, T.; Sakaguchi, Y. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3912. doi: 10.1073/pnas.1019480108
Liu, J.; Yu, H.; Li, S. M. Appl. Microbiol. Biotechnol. 2018, 102, 4435. doi: 10.1007/s00253-018-8908-6
Yao, T. T.; Liu, J.; Liu, Z. Z.; Li, T.; Li, H. Y.; Che, Q.; Zhu, T. J.; Li, D. H.; Gu, Q. Q.; Li, W. L. Nat. Commun. 2018, 9, 4091. doi: 10.1038/s41467-018-06411-x
(a) Li, S. M. ChemInform 2010, 27, 57.
(b) Xu, W.; Gavia, D. J.; Tang, Y. Nat. Prod. Rep. 2014, 31, 1474.
(c) Alkhalaf, L. M.; Ryan, K. S. Chem. Biol. 2015, 22, 317.
(d) Walsh, C. T. ACS Chem. Biol. 2014, 9, 2718.
Fan, A.; Winkelblech, J.; Li, S. M. Appl. Microbiol. Biotechnol. 2015, 99, 7399. doi: 10.1007/s00253-015-6813-9
(a) Meng, S.; Han, W.; Zhao, J.; Jian, X. H.; Pan, H. X.; Tang, G. L. Angew. Chem., Int. Ed. 2018, 57, 719.
(b) Patteson, J.; Cai, W.; Johnson, R. A.; Santa, M. K.; Li, B. Biochemistry 2017, 57, 61.
Kohn, H.; Widger, W. Curr. Drug Targets: Infect. Disord. 2005, 5, 273. doi: 10.2174/1568005054880136
Schmitt, E.; Bourgeois, G.; Gondry, M.; Aleksandrov, A. Sci. Rep. 2018, 8, 7031. doi: 10.1038/s41598-018-25479-5
Wunsch, C.; Mundt, K.; Li, S. M. Appl. Microbiol. Biotechnol. 2015, 99, 4213. doi: 10.1007/s00253-015-6490-8
Vior, N. M.; Lacret, R.; Chandra, G.; Dorairaj, S.; Trick, M. & Truman, A. W. Appl. Environ. Microbiol. 2018, 84(9), UNSP/ e02828-17.
Prasad, C. Peptides 1995, 16, 151. doi: 10.1016/0196-9781(94)00017-Z
Giessen, T. W.; von Tesmar, A. M.; Marahiel, M. A. Chem. Biol. 2013, 20, 828. doi: 10.1016/j.chembiol.2013.04.017

图 2 CDPSs系统进化树分析
Figure 2 Phylogenetic analysis of selected CDPSs
The tree was generated using the PhyML program (v3.1) based on the maximum likelihood method. CDPSs' names consist of the corresponding accession numbers in the NCBI database and the host organisms in which they were found. The two main branches corresponding to the XYP and NYH subfamilies are shown in blue and red, respectively

图 3 CDPSs晶体结构的比较
Figure 3 Comparison of the crystal structures of CDPSs
(A) Superimposition of the cartoon structures of AlbC (PDB ID, 3OQV; light magenta), Rv2275 (PDB ID, 2X9Q; green), and YvmC (PDB ID, 3OQH; cyan). (B) Superimposition of the cartoon structures of AlbC and TyrRS of M. jannaschii (PDB ID, 1J1U). AlbC is in light magenta; TyrRS is in lemon; a C-terminal domain involved in tRNA-binding and anticodon recognition coloured in gray. (C) Superimposition of the cartoon structures of AlbC and Rgry-CDPS (PDB ID, 5MLP). The first half of the Rossmann fold of AlbC is in hot pink and Rgry-CDPS is in light blue; the second half of the Rossmann fold of AlbC is in light magenta and Rgry-CDPS is in sky blue

图式 1 AlbC催化产生cyclo-(L-Phe-L-Phe)的分子机制[37c]
Scheme 1 Molecular mechanism of cyclo-(L-Phe-L-Phe) formation catalyzed by AlbC

图 5 Albonoursin的生物合成途径及其生物合成基因簇
Figure 5 Biosynthetic pathways and gene clusters of albonoursin

图 6 Pulcherrimin的生物合成途径及其生物合成基因簇
Figure 6 Biosynthetic pathways and gene clusters of pulcherrimin

图 7 Mycocyclosin的生物合成途径及其生物合成基因簇
Figure 7 Biosynthetic pathways and gene clusters of mycocyclosin

图 8 Nocazine family的生物合成途径及其生物合成基因簇
Figure 8 Biosynthetic pathways and gene clusters of nocazine family
The function of gene Ndas_1145 remains to be clarified

图 9 Bicyclomycin的生物合成途径及其生物合成基因簇
Figure 9 Biosynthetic pathways and gene clusters of bicyclomycin

图 10 Drimentines类化合物的生物合成途径及其生物合成基因簇
Figure 10 Biosynthetic pathways and gene clusters of drimentines
表 1 CDPSs表面结合口袋P1、P2内氨基酸残基序列比对结果a
Table 1. Specificity groups identified and their consensus motifs of P1 and P2 determined from characterized CDPSs
| Specificity group (*) | Consensus motif for P1 | Consensus motif for P2 |
| cLL (NYH) (7/123) | |
|
| cWW (NYH) (8/22) | |
|
| cCC (NYH) (8/60) | |
|
| cAE (XYP) (8 / 55) | |
|
| cXE (XYP) (9/81) | |
|
| cAX (XYP) (10/88) | |
|
| a The numbers in brackets (*) correspond to the number of CDPSs experimentally characterized vs. the total number of CDPSs in the group (Last up-date: June 2017, Gondry et al. 2018)[40]; degenerated positions are indicated in gray. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
