图 1.
Armchair结构焦炭模型(Am)
Figure 1.
Char model with armchair configuration (Am)

氮氧化物(NOx)会引发环境问题并影响人体健康。大型燃煤电站锅炉排放的NOx主要成分为NO,NO的有效控制与减排已成为能源利用与环境保护共同关注的问题[1]。已有大量工作致力于明确燃煤过程中NO生成与还原的反应机理并开发出多种NO控制技术[2, 3]。其中,再燃技术因脱硝效率高、投资成本低等优点,是目前应用最广泛的NOx控制技术之一[4, 5]。当以煤或生物质作为再燃燃料时,NO可在炉内高温热解产生的焦炭表面发生非均相还原[6-8]。焦炭非均相还原在总的NO还原中占比可高达59%[9]。此外,研究人员发现焦炭中的金属元素对NO的非均相还原有较大影响,尤其是在煤和生物质中含量较高的碱金属和碱土金属[10-13]。
一般认为,焦炭中的金属元素有利于NO的还原,但其详细作用机理非常复杂,至今尚未明确。Wang等[12]认为,碱金属元素通过类“氧化-还原”循环过程作用于NO的非均相还原反应。Zhang等[14]和周昊等[15]的实验研究认为,碱金属元素能够显著降低反应活化能,从而促进焦炭对NO的还原。Sörensen等[16]则发现,碱金属元素对反应活化能影响不大,但NO在原煤焦上的非均相还原速率比在脱灰煤焦上快2.5倍,作者分析认为,金属元素通过增加焦炭表面的反应活性位点加快反应的进行。Wu等[17]认为,碱金属对NO非均相还原的促进作用主要表现为碱金属会促进NO在焦炭表面的化学吸附,并提出了两种化学吸附结合方式,但无法确定哪种方式是主要的。目前,对金属元素的详细作用机理缺乏深入理解,故对一些复杂甚至矛盾的实验现象不能给出合理解释[18],限制了燃烧过程中NO控制措施的实施和新型低NOx燃烧技术的开发。因此,揭示金属元素对NO非均相还原的作用机理对煤炭清洁高效利用有重要意义。
受限于焦炭非均相反应的复杂性,部分数据和机理分析难以通过实验手段获得。分子模拟为反应机理的研究提供了另一种手段,并已用于研究煤的结构性质[19]、热解、燃烧和气化反应[20-25]。本研究拟在前期研究工作的基础上,采用密度泛函理论(Density Functional Theory, DFT)与波函数理论相结合的方法研究Na对NO非均相还原的作用机理。基于波函数理论分析分子体系的电子结构,预测NO分子的吸附位点;探寻非均相还原反应路径,通过热力学和动力学分析明确Na对NO吸附和非均相还原的影响;力求在分子水平揭示Na对NO非均相还原的微观作用机理。
煤及焦炭在三维空间内表现为短程有序、长程无序的非均一芳香团簇堆积结构[19, 26-28]。目前,采用DFT开展焦炭相关化学反应的研究时,广泛采用的模型主要有团簇模型和周期性石墨烯模型,两种模型在研究化学反应时各有特点,并取得了与实验符合较好的结果[22, 24, 29-32]。本研究选用团簇焦炭模型。焦炭边缘主要有“armchair”和“zigzag”两种构型。不同边缘构型的模型表现出不同的电子结构特性,进而展现出不同的反应性[33]。研究表明,zigzag边缘的焦炭具有更高的活性,但He等[34]通过像差校正透射电镜观测到当温度高于600℃时,焦炭边缘主要表现为armchair构型。Chen等[35]通过计算发现由4-6个苯环组成的芳香团簇模型可以很好地再现实验结果或解释实验现象,大规模扩大模型体系对计算结果并无明显改善。文献[24, 29-32]选用5-7个苯环组成的芳香平面模型研究焦炭参与的非均相反应并很好地解释了反应机理。因此,本研究选用具有armchair构型的焦炭模型(图 1),对边缘部分碳原子编号以便后续分析。
采用B3LYP交换相关泛函[36, 37],选用6-31G(d, p)基组优化几何结构,寻找过渡态与中间体。对优化所得结构全部进行频率计算和波函数稳定性测试,并对计算所得过渡态进行内禀反应坐标分析(IRC)[38]以确保过渡态与中间体的正确连接。在相同水平下计算单点能,并进行零点能校正。为减小频率谐振近似引起的误差,在计算零点能时考虑热校正因子[39]。文献[33, 40]指出在模拟碳基材料体系时不应忽视色散作用的影响。B3LYP泛函对相关势的长程描述不够准确,不能很好地描述色散作用,但结合Grimme提出的DFT-D3(BJ)方法[41]可以非描述色散作用,因此,本研究在所有计算中均考虑色散校正。量子化学计算在Gaussian09程序包[42]上进行。
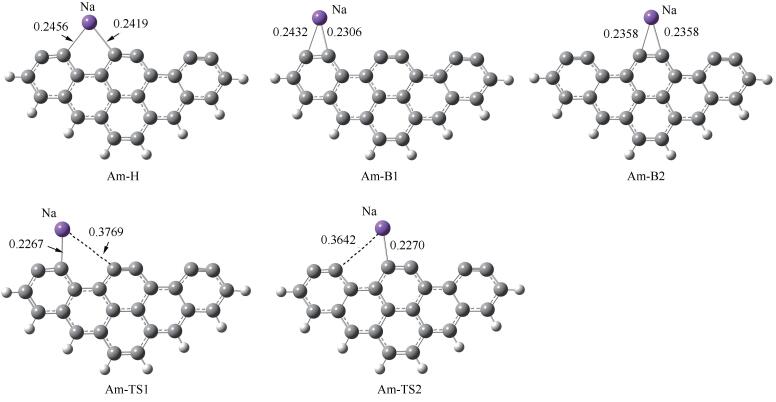
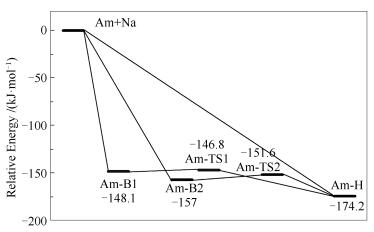
煤中钠主要以有机和无机两种形态存在,高温时钠会以Na原子、NaCl等形式挥发到气相中[43-45]。在模拟金属元素对煤焦相关反应影响时,通常采用向焦炭模型中修饰金属原子的方法[24, 32, 46-49]。Armchair模型的电子结构分析显示孤对电子主要分布在边缘的不饱和碳原子周围[33],可推测Na原子倾向于吸附在焦炭边缘,与文献[50, 51]的结论相符。因此,本研究重点关注Na原子在Am边缘的吸附。对Na在边缘穴位、边缘桥位上的吸附进行模拟,优化得到三个稳定构型(图 2)。图 3为Na吸附的势能面图(Potential Energy Surface, PES)。Na在Am边缘的吸附为放热反应,吸附在边缘穴位释放出最多能量,为174.2kJ/mol。Am-B1和Am-B2可分别经由Am-TS1和Am-TS2仅克服1.3和5.4kJ/mol的能垒转化为Am-H,说明Na原子可以在焦炭边缘游离,但从热力学角度最倾向于稳定在边缘穴位,因此,本研究重点分析Am-H的电子结构,后续NO分子的吸附与非均相还原的模拟也基于Am-H结构展开。
电子结构分析可以更深入地揭示分子体系的反应性并可预测反应位点[52, 53]。因此,本研究提取了Am-H的波函数信息,并将其电子结构可视化。图 4(a)给出了Am-H的电子密度差,红色实线表示区域电子密度增加,黑色虚线表示电子密度减少。由图 4(a)可知,Na原子外围电子密度减少,说明Na失电子。Na与边缘碳原子之间有电子密度增大的区域,但电子结构特性表现为非共价键。为研究Na原子与边缘碳原子之间的作用,进行了约化密度梯度函数(Reduced Density Gradient, RDG)[54]分析。图 4(b)给出了RDG为0.5的等值面图、图 4(c)为RDG散点图,横坐标为电子密度Hessian矩阵第二大本征值sign(λ2)与电子密度ρ的乘积,纵坐标为RDG函数值。蓝色表示强吸引作用,绿色表示范德华作用,红色表示互斥作用。RDG分析显示Na和两个碳原子之间表现为强的静电吸引作用,生成的五元环中存在环张力。为进一步明确Na原子与C3、C6原子的成键特性,计算并分析了Am-H的电子定域化函数(Electron Localization Function, ELF),具体见图 4(d)。区域ELF数值越高,电子定域性越强;反之越弱。可以看出,由于Na原子和C3、C6原子之间主要靠强静电力束缚,相互间没有共价作用,原子间ELF函数值极低,Na原子上的外壳电子几乎被C3和C6原子全部吸走而形成满壳层的Na+。图 4(e)考察了Am-H的Hirshfeld电荷分布,发现电子由Na原子转移到焦炭上。与文献[33]对比可知,Na吸附后,焦炭边缘的碳原子带有更多的负电荷。图 4(f)给出了范德华(van der Waals, vdW)分子表面静电势(Electrostatic potential, ESP)分布和静电势极值点分布,可以看出,Na吸附在焦炭上后,体系静电势分布更加不均匀[33],Na原子外围分子表面主要表现为正静电势,存在静电势最大值463.3kJ/mol;C7和C10原子之间的边缘穴位存在静电势极小值-72.1kJ/mol。计算得到Am-H的电负性为3.24eV,文献[33]中Am的电负性为3.92eV,NO分子的电负性为7.74eV。体系电负性越低,给电子能力越强。分析可得以下结论:NO向Am-H的化学吸附为亲电反应;相比Na原子,NO更倾向于与边缘带有负电荷的碳原子结合,Jiao等[48]也发现,边缘碳活性位点对NO的吸引力比焦炭边缘的金属位点更强;Na的掺杂促进NO在焦炭边缘的吸附。
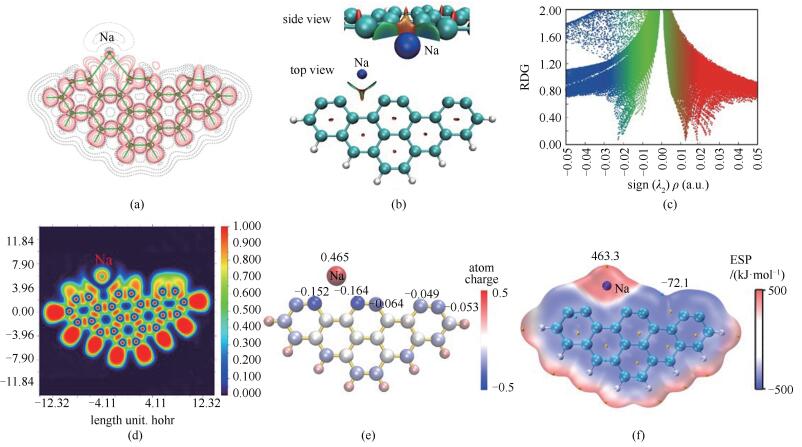
(a): electron density difference plots; (b): iso-surface of RDG (isovalue 0.5); (c): scatter graph of RDG vs sign(λ2)ρ; (d): color-mapped ELF; (e): distribution of Hirshfeld atomic charge; (f): ESP mapped vdW surface
基于NO最倾向于以side-on模式吸附在焦炭边缘[22, 31, 33, 35, 55, 56],对NO分子在Am-H边缘上的两种side-on吸附进行模拟,优化得到两个吸附结构(图 5)。自由气体状态下NO分子中的N-O键长为0.116nm,吸附后N-O键被明显拉长,两种吸附分别释放出271.8和250kJ/mol的能量。比较可知,NO中的O临近Na原子的吸附方式释放出更多的热量。从热力学角度AmNaNO-A更稳定。因此,本研究重点分析AmNaNO-A的电子结构,后续NO非均相还原的模拟也基于AmNaNO-A结构展开。文献[55]中NO以side-on模式吸附在armchair焦炭边缘释放出198.5kJ/mol的能量,可见Na促进第一个NO分子的吸附。
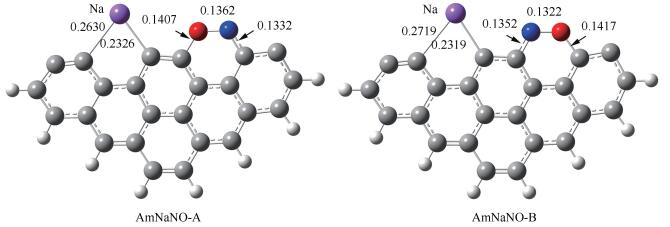
由图 6(a)中AmNaNO-A的电子密度差分布可知,NO吸附后,Na原子周围的电子密度进一步减少。Na原子与C3之间的电子密度明显减少,说明NO的吸附减弱了C3-Na键,C3-Na键的键长由Am-H中的0.2456nm伸长至AmNaNO-A中的0.2630nm。C11原子外围电子密度显著增多,说明该区域存在未成对电子。图 6(b)给出了vdW分子表面ESP分布和ESP极值点分布,与Am-H(图 4(f))相比,NO吸附后,体系静电势分布更不均匀,Na原子外围分子表面存在静电势最大值490.7kJ/mol;C11原子外围分子表面存在静电势最小值-253.3kJ/mol。计算得到AmNaNO-A的电负性为3.69eV,则后续第二个NO分子的吸附亦为亲电反应,推测其最倾向于吸附在AmNaNO-A静电势极小值点。
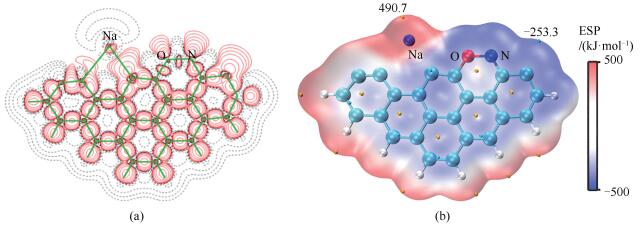
(a): electron density difference plots; (b): ESP mapped vdW surface
已有文献[22, 55, 56]详尽研究了NO在纯碳边缘的非均相还原路径,一致认为N2的解吸附步为非均相还原反应速率控制步,因此,本研究对NO在纯碳边缘非均相还原的整个路径不再赘述,但为了便于计算结果的对比,在此给出NO在Am边缘非均相还原中N2解吸附的基元反应步。N2解吸附基元反应步所涉及的几何结构如图 7所示。Am-M→Am-P+N2基元反应活化能为144.7kJ/mol。
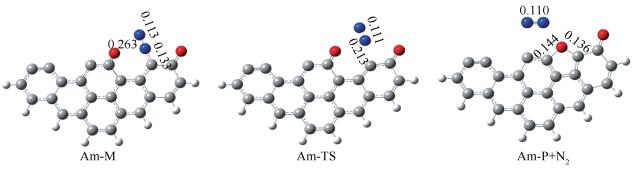
下面重点结合2.2.2节反应位点的预测结果探寻NO在含Na焦炭边缘的非均相还原通道。优化所得反应通道中的中间体、过渡态和产物的几何构型见图 8,同时标注了部分键长数据。图 9为非均相还原的PES图和通道中各结构的RDG图。
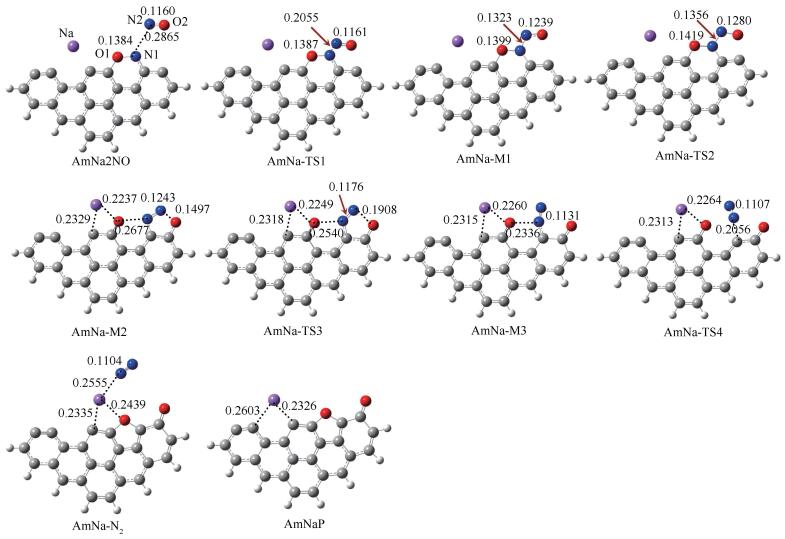
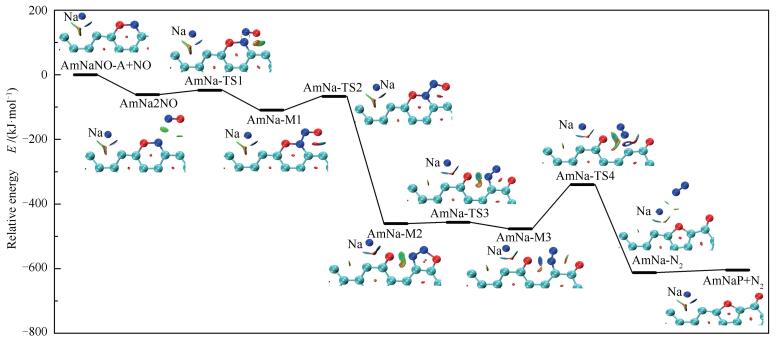
首先第二个NO分子在预测的吸附位点与AmNaNO-A结合生成复合物AmNa2NO,释放出61.3kJ/mol的能量;结合图 9中的RDG图可以看出,N1与N2之间、O2与C11之间存在范德华作用力。AmNa2NO需克服13.1kJ/mol的能垒经由AmNa-TS1生成中间体AmNa-M1。在之前的研究中,均未关注此基元反应,而认为第二个NO分子的吸附是无势垒自发过程[48, 55, 56]。AmNaNO-A+NO→AmNaM1共释放出109.2kJ/mol的能量,Zhao等[56]计算得到第二个NO分子在纯碳边缘的吸附释放出100kJ/mol的能量,可见Na对第二个NO分子的吸附影响不大。由AmNa-M1的RDG图观察到O2原子和C11原子之间存在强的吸引力,因此在下一步翻越42.2kJ/mol的能垒生成O2-C11键,同时伴随着O1-N1键的断裂和Na原子的游离。AmNa-M2的RDG等值面显示O1与N1之间表现为范德华力;Na与C3原子之间的作用力消失,Na与O1之间表现出强的静电吸引力。AmNa-M1→AmNa-M2为强放热过程,释放出351.3kJ/mol的能量。随后N2-O2键断裂,生成开环结构AmNa-M3,该步反应活化能仅为3.9kJ/mol。最终C10-N1键翻越136.5kJ/mol的能垒、经AmNa-TS4断裂,同时伴随含氧五元环的生成,生成复合物AmNa-N2。复合物AmNa-N2的RDG图显示N1与O1、Na之间存在范德华作用力,Na与O1原子之间由强静电吸引力减弱为范德华作用力。AmNa-N2可以看作N2在焦炭边缘的物理吸附产物,N2分子挣脱范德华力的束缚释放出去需吸收7.9kJ/mol的能量。N2分子释放后,Na原子重新游离回最初的边缘穴位。非均相还原反应通道中C10-N1键的断裂为最高能垒步,即N2的化学解吸附步,整个反应AmNaNO-A+NO→AmNaP+N2释放出604.4kJ/mol的能量。
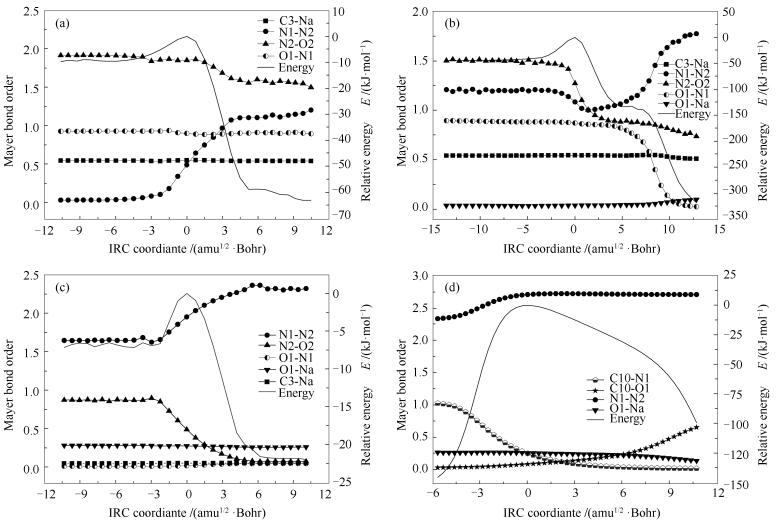
为定量判断反应过程中原子间的成断键,对反应通道中的各个过渡态进行IRC计算,并对IRC路径上的每个结构进行单点计算,提取各结构中原子间的Mayer键级[57],进而得到整个反应通道中各化学键的Mayer键级变化,具体见图 10,横坐标0点代表过渡态结构。
(a): AmNa-TS1; (b): AmNa-TS2; (c): AmNa-TS3; (d): AmNa-TS4
由图 10(a)可知,经历TS1,N1-N2的键级由0显著增大至1.23,N2-O2的键级由1.9降至1.5,定量反映出N1-N2键的生成与N2-O2键的减弱;O1-N1和C3-Na的键级保持稳定。经历TS2,N1-N2的键级进一步增大至1.7;N2-O2的键级降低至0.7;O1-N1的键级在该基元反应中前期保持稳定,后期迅速下降至0;将N2-O2、O1-N1的键级变化与IRC路径的能量变化结合来看,可以看出,经历TS2后沿反应前进的方向,能量的降低主要受N2-O2键减弱的影响,经历一个小的平台能量继续迅速下降则主要归因于O1-N1键的断裂;此外,在反应后期可以观察到C3-Na的键级略降,对应O1-Na的键级略升,说明Na原子逐渐向O1原子游离。随后经历TS3,N2-O2键级降至0,N1-N2键级则继续增长至2.3,说明N2-O2键的断裂与N1-N2键的增强;C3-Na的键级维持在接近于0的水平,说明Na与C3原子之间的作用力消失;而O1-Na的键级稳定在0.3左右,意味着Na原子与O1原子之间形成稳定的作用力。最后经历TS4,N1-N2键级继续升高至2.71,N2分子自由状态下N-N键级为2.74,表明非均相还原生成的N2分子结构已基本稳定;C10-N1键级持续降低至0,意味着N2分子的化学解吸附;同时C10-O1键级不断增长,表明五元环逐步形成;反应后期O1-Na键级出现减小的趋势,意味着两者之间作用力减弱,对应图 8和图 9中Na原子重新游离回边缘穴位。由以上分析可以看出,Mayer键级确实可定量反映化学键的生成与断裂,更直观地表现出反应过程中原子间相互作用的变化关系,对分析微观反应机理有重要意义。
经典过渡态理论(Conventional transition state theory, CTST)有很好的实用性,但CTST在推导过程中假定产物不会回穿过分界面返回到产物,从而高估了高温下的反应速率常数。为解决此问题,发展了变分过渡态理论(Variational transition state theory, VTST),其中,正则变分理论(Canonical variational theory, CVT)是使用最广的VTST方法[58]。采用Wigner方法考虑隧道效应,反应速率常数计算公式为:
|
$ k\left( T \right) = \kappa (T)\frac{{\sigma \;{Q^ \ne }\left( {T, {s_0}} \right)}}{{\beta h\;\;{\text{ }}{Q^{\text{R }}}(T)}}{\text{exp }}[ - \beta V({s_0})] $ |
(1) |
式中,σ为反应路径简并度;β=(kBT)-1,kB为Boltzmann常数;h为普朗克常数;T为温度;Q≠(T, s0)表示过渡态的配分函数(忽略虚频振动贡献);QR(T)为反应物配分函数;V(s0)表示势垒高度;κ(T)为Wigner透射系数;
|
$ \kappa \left( T \right) = 1 + \frac{1}{{24}}{\left[ {\frac{{hIm{\nu ^ \ne }}}{{{k_{\text{B}}}T}}} \right]^2} $ |
(2) |
式中,Im(ν≠)为过渡态虚频频率。
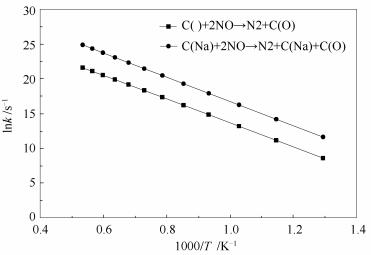
根据式(1)和(2)对NO在纯碳边缘和在Na修饰焦炭边缘非均相还原的速率控制步,即N2化学解吸附步进行计算,得到速控步在773-1873K温度范围内的速率常数,如图 11所示。拟合得到两个反应Arrhenius表达式的动力学参数见表 1,拟合所得活化能与Zhang等[14]的JC焦及Sörensen等的实验结果[16]符合较好。由图 11可知,在计算温度区间内,N2在添加Na焦炭边缘的化学解吸附速率高于在纯碳边缘的化学解吸附速率,说明Na可促进N2的化学解吸附从而加快NO的非均相还原。对比表 1中两个反应的动力学参数可知,Na的添加对速控步的活化能无明显影响,但指前因子显著增大。在过渡态理论中,指前因子与过渡态的活化熵有关,可反映出活化分子在全部分子中所占的比例[58],说明添加Na后焦炭边缘的活性位点增多,有利于加快NO非均相还原的进行。
 下载:
导出CSV
下载:
导出CSV
| Reaction | A/s-1 | Ea /(kJ·mol-1) |
| C()+2NO→N2+C(O) | 2.37×1013 | 148.5 |
| C(Na)+2NO→N2+C(Na)+C(O) | 7.04×1014 | 139.5 |
基于密度泛函理论与波函数理论探究了Na对焦炭非均相还原的微观作用机理;研究了Na在焦炭边缘的吸附行为,并提取稳定吸附结构的波函数信息分析电子结构、预测活性位点;进一步研究NO的吸附行为、探寻NO的非均相还原通道,基于Mayer键级变化定量分析关键化学键的成断键;综合热力学和动力学计算结果明确Na的作用机理。
Na原子在焦炭边缘的化学吸附为放热反应,最倾向于吸附在焦炭边缘的穴位,电子由Na转移到焦炭边缘的碳原子。
Na会增强第一个NO分子在焦炭边缘的吸附,但对第二个NO分子的吸附影响不大。
N2的化学解吸附步是非均相还原的速控步,Na的添加对速控步的活化能无明显影响,但依然会加快NO非均相还原的反应速率,主要原因是添加Na后焦炭上的活性位点增多。
Na原子通过在焦炭边缘的游离与相邻O原子之间发生“结合-分离”,通过类“氧化-还原”循环过程影响反应的进行。
王志轩, 刘志强. 我国煤电大气污染物控制现状及展望[J]. 中国工程科学, 2015,17,(9): 56-62. WANG Zhi-xuan, LIU Zhi-qiang. Current situation and prospect of control on air pollutants from coal-fired power in China[J]. Eng Sci, 2015, 17(9): 56-62.
CHANG S Y, ZHUO J K, MENG S, QIN S Y, YAO Q. Clean coal technologies in China:Current status and future perspectives[J]. Eng, 2016, 2: 447-459.
GLARBORG P, MILLER J A, RUSCIC B, KLIPPENSTEIN S J. Modeling nitrogen chemistry in combustion[J]. Prog Energy Combust Sci, 2018, 67: 31-68.
SMOOT L D, HILL S C, XU H. NOx control through reburning[J]. Prog Energy Combust Sci, 1998, 24(5): 385-408.
刘艳华, 张晓燕, 刘银河, 车得福. 再燃煤粉的NO还原特性研究[J]. 燃料化学学报, 2007,35,(5): 523-527. LIU Yan-hua, ZHANG Xiao-yan, LIU Yin-he, CHE De-fu. NO reduction behavior of coal powder used for reburning[J]. J Fuel Chem Technol, 2007, 35(5): 523-527.
钟北京, 施卫伟, 傅维标. 煤粉再燃过程中NO异相还原机理的重要性[J]. 燃烧科学与技术, 2002,8,(1): 6-8. ZHONG Bei-jing, SHI Wei-wei, FU Wei-biao. Importance of heterogeneous mechanisms of NO reduction during reburning with pulverized char[J]. J Combust Sci Technol, 2002, 8(1): 6-8.
张超群, 姜秀民, 黄庠永, 刘建国. 煤焦吸附NO特性与红外光谱分析[J]. 化工学报, 2007,58,(3): 581-586. ZHANG Chao-qun, JIANG Xiu-min, HUANG Xiang-yong, LIU Jian-guo. Characteristics of adsorption of NO gas on coal char and FTIR analysis[J]. CIESC J, 2007, 58(3): 581-586.
GUO F, Wu R C, BAXTER L L, HECKER W C. Models to predict kinetics of NOx reduction by chars as a function of coal rank[J]. Energy Fuels, 2019, 33: 5498-5504.
王永桥, 陆飞, 刘永生, 卢平. 生物质再燃脱硝及异相还原研究[J]. 中国电机工程学报, 2010,30,(26): 101-106. WANG Yong-qiao, LU Fei, LIU Yong-sheng, LU Ping. Study on NOx reduction and its heterogeneous mechanism during biomass reburning[J]. Proc CSEE, 2010, 30(26): 101-106.
赵宗彬, 李文, 李保庆. 矿物质对煤焦燃烧过程中NO释放规律的影响[J]. 化工学报, 2003,54,(1): 100-106. ZHAO Zong-bin, LI Wen, LI Bao-qing. Effect of mineral matter on release of NO during coal char combustion[J]. CIESC J, 2003, 54(1): 100-106.
ZHONG B J, TANG H. Catalytic NO reduction at high temperature by de-ashed chars with catalysts[J]. Combust Flame, 2007, 149: 234-243.
WANG Z H, ZHOU J H, WEN Z C, LIU J Z, CEN K F. Effect of mineral matter on NO reduction in coal reburning process[J]. Energy Fuels, 2007, 21(4): 2038-2043.
信晶, 尹书剑, 孙保民, 朱恒毅, 罗肖, 黄强, 肖海平. 掺杂金属化合物强化焦炭-NO反应的析因试验研究[J]. 煤炭学报, 2015,40,(5): 1174-1180. XIN Jing, YIN Shu-jian, SUN Bao-min, ZHU Heng-yi, LUO Xiao, HUANG Qiang, XIAO Hai-ping. Factorial experimental study of Analysis of the char-NO reaction intensified by doped metallic compounds[J]. J China Coal Soc, 2015, 40(5): 1174-1180.
ZHANG J W, SUN S Z, ZHAO Y J, HU X D, XU G W, QIN Y K. Effects of inherent metals on NO reduction by coal char[J]. Energy Fuels, 2011, 25: 5605-5610.
周昊, 刘瑞鹏, 刘子豪, 程明, 岑可法. 碱金属对焦炭燃烧过程中NOx释放的影响[J]. 煤炭学报, 2015,40,(5): 1160-1164. ZHOU Hao, LIU Rui-peng, LIU Zi-hao, CHENG Ming, CEN Ke-fa. Influence of alkali metal on the evolution of NOx during coke combustion[J]. J China Coal Soc, 2015, 40(5): 1160-1164.
SØRENSEN C O, JOHNSSON J E, JENSEN A. Reduction of NO over wheat straw char[J]. Energy Fuels, 2001, 15: 1359-1368.
WU X Y, SONG Q, ZHAO H B, YAO Q. Catalytic mechanism of inherent potassium on the char-NO reaction[J]. Energy Fuels, 2015, 29(11): 7566-7571.
吕俊复, 柯希玮, 蔡润夏, 张缦, 吴玉新, 杨海瑞, 张海. 循环流化床燃烧条件下焦炭表面NOx还原机理研究进展[J]. 煤炭转化, 2018,41,(1): 1-12. LV Jun-fu, KE Xi-wei, CAI Run-xia, ZHANG Man, WU Yu-xin, YANG Hai-rui, ZHANG Hai. Research progress on the kinetics of NOx reduction over chars in fluidized bed combustion[J]. Coal Convers, 2018, 41(1): 1-12.
周星宇, 曾凡桂, 相建华, 邓小鹏, 相兴华. 马脊梁镜煤有机质大分子模型构建及分子模拟[J]. 化工学报, 2020,71,(4): 1802-1811. ZHOU Xing-yu, ZENG Fan-gui, XIANG Jian-hua, DENG Xiao-peng, XAING Xing-hua. Macromolecular model construction and molecular simulation of organic matter in Majiliang vitrain[J]. CIESC J, 2020, 71(4): 1802-1811.
ZHENG M, PAN Y, WANG Z, LI X X, GUO L. Capturing the dynamic profiles of products in Hailaer brown coal pyrolysis with reactive molecular simulations and experiments[J]. Fuel, 2020, 268: 117290.
杨慧芳, 关海莲, 李平, 夏英, 王凤, 徐文静, 白红存, 郭庆杰. 煤颗粒燃烧过程氧化机理及有机氮转化的分子模拟:以宁东红石湾煤为例[J]. 化工学报, 2020,71,(2): 799-810. YANG Hui-fang, GUAN Hai-lian, LI Ping, XIA Ying, WANG Feng, XU Wen-jing, BAI Hong-cun, GUO Qing-jie. Molecular modeling of oxidation mechanism and organic nitrogen conversion in coal particle combustion:A case study on HSW coal of Ningdong[J]. CIESC J, 2020, 71(2): 799-810.
许紫阳, 岳爽, 王春波, 刘瑞琪. 焦炭催化CO还原NO的反应机理研究[J]. 燃料化学学报, 2020,48,(3): 266-274. XV Zi-yang, YUE Shuang, WANG Chun-bo, LIU Rui-qi. Reaction mechanism of NO reduction with CO catalyzed by char[J]. J Fuel Chem Technol, 2020, 48(3): 266-274.
王新伟, 马强, 韩超杰, 林日亿. 四氢噻吩与硫酸镁热化学还原生成H2S机制[J]. 中国石油大学学报(自然科学版), 2020,44,(1): 156-162. WANG Xin-wei, MA Qiang, HAN Chao-jie, LIN Ri-yi. Study on formation mechanism of H2S by thermochemical sulfate reduction of thiolane and magnesium sulphate[J]. J China Univ Pet, 2020, 44(1): 156-162.
ZHAO D, LIU H, SUN C, XU L, CAO Q. DFT study of the catalytic effect of Na on the gasification of carbon-CO2[J]. Combust Flame, 2018, 197: 471-86.
陈玉, 张福丽, 姚辉超, 刘植昌, 崔佳, 徐春明. 催化水煤气变换反应的计算模拟进展[J]. 化工进展, 2012,31,(10): 2221-2227. CHEN Yu, ZHANG Fu-li, YAO Hui-chao, LIU Zhi-chang, CUI Jia, XU Chun-ming. Progress of theoretical simulation of catalytic water-gas-shift reaction[J]. Chem Ind Eng Prog, 2012, 31(10): 2221-2227.
PERRY S T, HAMBLY E M, FLETCHER T H, SOLUM M S, PUGMIRE R J. Solid-state 13C NMR characterization of matched tars and chars from rapid coal devolatilization[J]. Proc Combust Inst, 2000, 28: 2313-2319.
SHENG C. Char structure characterised by Raman spectroscopy and its correlations with combustion reactivity[J]. Fuel, 2007, 86: 2316-2324.
王宝俊, 章丽娜, 凌丽霞, 章日光. 煤分子结构对煤层气吸附与扩散行为的影响[J]. 化工学报, 2016,67,(6): 2548-2557. WANG Bao-jun, ZHANG Li-na, LING Li-xia, ZHANG Ri-guang. Effects of coal molecular structure on adsorption and diffusion behaviors of coalbed methane[J]. CIESC J, 2016, 67(6): 2548-2557.
赵鹏飞, 郭欣, 郑楚光. 活性炭及氯改性活性炭吸附单质汞的机制研究[J]. 中国电机工程学报, 2010,30,(23): 40-44. ZHAO Peng-fei, GUO Xin, ZHENG Chu-guang. Investigation the mechanism of elemental mercury binding on activated carbon and chlorine-embedded activated carbon[J]. Proc CSEE, 2010, 30(23): 40-44.
张秀霞, 谢苗, 伍慧喜, 吕晓雪, 林日亿, 周志军. 钙对焦炭非均相还原NO的微观作用机理:DFT研究[J]. 燃料化学学报, 2020,48,(2): 163-171. ZHANG Xiu-xia, XIE Miao, WU Hui-xi, LÜ Xiao-xue, LIN Ri-yi, ZHOU Zhi-jun. Microscopic effect mechanism of Ca on NO heterogeneous reduction by char:A DFT study[J]. J Fuel Chem Technol, 2020, 48(2): 163-171.
陈萍, 顾明言, 汪嘉伦, 卢坤, 林郁郁. 含氮煤焦还原NO反应路径研究[J]. 燃料化学学报, 2019,47,(3): 279-286. CHEN Ping, GU Ming-yan, WANG Jia-lun, LU Kun, LIN Yu-yu. Reaction pathways for the reduction of NO by nitrogen-containing char[J]. J Fuel Chem Technol, 2019, 47(3): 279-286.
高正阳, 刘晓硕, 李昂, 马传志, 李祥, 杨建蒙. 电厂烟气中SO2对活性炭吸附单质铅(Pb0)的影响机理[J]. 环境科学学报, 2019,39,(11): 3732-3739. GAO Zheng-yang, LIU Xiao-shuo, LI Ang, MA Chuan-zhi, LI Xiang, YANG Jian-meng. The effect of SO2 on adsorption of element lead toward activated carbon in coal-fired power plants[J]. Acta Sci Circums, 2019, 39(11): 3732-3739.
ZHANG X X, WU H X, XIE M, LÜ X X, ZHOU Z J, LIN R Y. Wave function and molecular reactivity study of char with different edges and the chemisorption properties of nitric oxide[J]. J Energy Inst, 2020, 93(4): 1519-1526.
HE K, ROBERTSON A W, FAN Y, ALLEN C H, LIN Y C, SUENAGA K, KIRKLAND A I, WARNER J H. Temperature dependence of the reconstruction of zigzag edges in graphene[J]. ACS Nano, 2015, 9(5): 4786-95.
CHEN N, YANG R T. Ab initio molecular orbital calculation on graphite:Selection of molecular system and model chemistry[J]. Carbon, 1998, 36: 1061-1070.
STEPHENS P, DEVLIN F, CHABALOWSKI C, FRISCH M. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields[J]. J Phys Chem, 1994, 98: 11623-11627.
吕泽康, 龙慎伟, 李冠兵, 牛胜利, 路春美, 韩奎华, 王永征. 生物质锅炉氯腐蚀的密度泛函理论研究[J]. 化工学报, 2019,70,(11): 4370-4376. LV Ze-kang, LONG Shen-wei, LI Guan-bing, NIU Sheng-li, LU Chun-mei, HAN Kui-hua, WANG Yong-zheng. Density functional theory study on chlorine corrosion of biomass furnace[J]. CIESC J, 2019, 70(11): 4370-4376.
GONZALEZ C, SCHLEGEL H B. Reaction path following in mass-weighted internal coordinates[J]. J Phys Chem, 1990, 94: 5523-5527.
MERRICK J P, MORAN D, RADOM L. An evaluation of harmonic vibrational frequency scale factors[J]. J Phys Chem A, 2007, 111: 11683-11700.
BURSCH M, CALDEWEYHER E, HANSEN A, NEUGEBAUER H, EHLERT S, GRIMME S. Understanding and quantifying london dispersion effects in organometallic complexes[J]. Acc Chem Res, 2019, 52: 258-266.
GOERIGK L, HANSEN A, BAUER C, EHRLICH S, NAJIBI A, GRIMME S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions[J]. Phys Chem Chem Phys, 2017, 19: 32184-32215.
FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09[CP]. Revision D.01; Gaussian, Inc., Wallingford CT, 2009.
张守玉, 陈川, 施大钟, 吕俊复, 王健, 董爱霞, 熊绍武.高钠煤燃烧利用现状[J].中国电机工程学报, 2013, 33(5): 1-12.ZHANG Shou-yu, CHEN Chuan, SHI Da-zhong, LÜ Jun-fu, WANG Jian, DONG Ai-xia. Situation of combustion utilization of high sodium coal[J]. Proc CSEE, 2013, 33(5): 1-12.
宋维健, 宋国良, 齐晓宾, 吕清刚. 准东高钠煤气化过程中Na的迁移转化规律[J]. 煤炭学报, 2016,41,(2): 490-496. SONG Wei-jian, SONG Guo-liang, QI Xiao-bin, LÜ Qing-gang. Sodium transformation law of Zhundong coal during gasification[J]. J China Coal Soc, 2016, 41(2): 490-496.
魏砾宏, 崔保崇, 陈勇, 杨天华, 郭良振. 高碱煤钠赋存形态及其燃烧过程中迁移转化的研究进展[J]. 燃料化学学报, 2019,47,(8): 897-906. WEI Li-hong, CUI Bao-chong, CHEN Yong, YANG Tian-hua, GUO Liang-zhen. Occurrence of sodium in high alkali coal and its transformation during combustion[J]. J Fuel Chem Technol, 2019, 47(8): 897-906.
刘磊, 金晶, 林郁郁, 侯封校. 钙元素对焦炭表面NO吸附行为的影响:密度泛函理论研究[J]. 燃料化学学报, 2015,43,(12): 1414-1419. LIU Lei, JIN Jing, LIN Yu-yu, HOU Feng-xiao. Effect of calcium on the absorption of NO on char surface:A density functional theory study[J]. J Fuel Chem Technol, 2015, 43(12): 1414-1419.
刘吉, 陆强, 蒋晓燕, 胡斌, 董长青, 杨勇平. 碱金属离子对吡咯热解生成NOx前驱物HCN机理的影响[J]. 煤炭学报, 2018,43,(9): 2633-2638. LIU Ji, LU Qiang, JIANG Xiao-yan, HU Bin, DONG Chang-qing, YANG Yong-ping. Effect of alkali metal ions on the formation mechanism of HCN as NOx precursor during pyrrole pyrolysis[J]. J China Coal Soc, 2018, 43(9): 2633-2638.
JIAO A Y, JIANG X M, LIU J X. Density functional theory investigation on the catalytic reduction of NO by CO on the char surface:the effect of iron[J]. Environ Sci Technol, 2020, 54: 2422-2428.
ZHANG X X, XIE M, WU H X, LÜ X X, Zhou Z J. DFT study of the effect of Ca on NO heterogeneous reduction by char[J]. Fuel, 2020, 265: 116995.
MPOURNPAKIS G, FROUDAKIS G. Why alkali metals preferably bind on structural defects of carbon nanotubes:A theoretical study by first principle[J]. J Chem Phys, 2006, 125: 204707.
FAROKH N A H, ROMAN T, HUSSAIN T, SEARLES D J. Computational study on the adsorption of sodium and calcium on edge-functionalized graphene nanoribbons[J]. J Phys Chem C, 2019, 123: 14895-14908.
LU T, CHEN F W. Multiwfn:A multifunctional wavefunction analyzer[J]. J Comput Chem, 2012, 33: 580-592.
邓军, 李亚清, 张玉涛, 杨超萍, 张静, 史学强. 羟基(-OH)对煤自燃侧链活性基团氧化反应特性的影响[J]. 煤炭学报, 2020,45,(1): 232-240. DENG Jun, LI Ya-qing, ZHANG Yu-tao, YANG Chao-ping, ZHANG Jing, SHI Xue-qiang. Effects of hydroxyl on oxidation characteristics of side chain active groups in coal[J]. J China Coal Soc, 2020, 45(1): 232-240.
JOHNSON E R, KEINAN S, MORI-SÁNCHEZ P, CONTRERAS-GARCÍA J, COHEN A J, YANG W T. Revealing noncovalent interactions[J]. JACS, 2010, 132: 6498-6506.
张秀霞, 周志军, 周俊虎, 刘建忠, 岑可法. 煤粉再燃中煤焦异相还原NO机理的量化研究[J]. 燃烧科学与技术, 2011,17,(2): 155-159. ZHANG Xiu-xia, ZHOU Zhi-jun, ZHOU Jun-hu, LIU Jian-zhong, CEN Ke-fa. A quantum chemistry study of heterogeneous reduction mechanisms of NO on the surface of char during pulverized coal reburning[J]. J Combust Sci Technol, 2011, 17(2): 155-159.
ZHAO T, SONG W L, FAN C G, LI S G, GLARBORG P, YAO X Q. Density functional theory study of the role of a carbon-oxygen single bond group in the NO-char reaction[J]. Energy Fuels, 2018, 32: 7734-7744.
MAYER I. Charge, Bond order and valence in the ab initio SCF theory[J]. Chem Phys Lett, 1983, 97: 270.
GARRETT B C, TRUHLAR D G. Accuracy of tunneling corrections to transition state theory for thermal rate constants of atom transfer reactions[J]. J Phys Chem, 1979, 83(1): 200-203.

图 2 Na在焦炭边缘的吸附结构(键长单位:nm)
Figure 2 Adsorption configurations of Na at the edge of char (bond length: nm)

图 3 Na在焦炭边缘吸附的反应势能面
Figure 3 Potential energy surface for adsorption of Na at the edge of char

图 4 Am-H电子结构分析
Figure 4 Electronic structure analysis of Am-H
(a): electron density difference plots; (b): iso-surface of RDG (isovalue 0.5); (c): scatter graph of RDG vs sign(λ2)ρ; (d): color-mapped ELF; (e): distribution of Hirshfeld atomic charge; (f): ESP mapped vdW surface

图 5 NO吸附产物几何结构(键长单位:nm)
Figure 5 Optimized adsorption configurations of the NO molecule (bond length: nm)

图 6 AmNaNO-A电子结构分析
Figure 6 Electronic structure analysis of AmNaNO-A
(a): electron density difference plots; (b): ESP mapped vdW surface

图 7 N2在Am边缘释放步中间体与过渡态的几何构型(键长单位:nm)
Figure 7 Geometrical parameters for stable species and the transition states for N2 desorption from Am (bond length: nm)

图 8 NO在Na修饰焦炭边缘非均相还原反应通道中的中间体与过渡态的几何构型(键长单位:nm)
Figure 8 Geometrical parameters for stable species and transition states in the pathway of NO heterogeneous reduction at the edge of Na-decorated char (bond length: nm)

图 9 NO非均相还原反应势能面
Figure 9 Potential energy surface of NO heterogeneous reduction on char

图 10 过渡态IRC路径上的键级变化
Figure 10 Mayer bond orders along IRC of transition states
(a): AmNa-TS1; (b): AmNa-TS2; (c): AmNa-TS3; (d): AmNa-TS4
表 1 拟合所得反应动力学参数
Table 1. Fitted kinetic parameters of Arrhenius expressions
| Reaction | A/s-1 | Ea /(kJ·mol-1) |
| C()+2NO→N2+C(O) | 2.37×1013 | 148.5 |
| C(Na)+2NO→N2+C(Na)+C(O) | 7.04×1014 | 139.5 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们