-
[1]
马璨, 吕迎春, 李泓. 储能科学与技术, 2014, 3(1):53~65. doi: 10.3969/j.issn.2095-4239.2014.01.008
-
[2]
田志宏, 赵海雷, 王治峰, 等.电池, 2008, 38(3):186~188. doi: 10.3969/j.issn.1001-1579.2008.03.019
-
[3]
程科, 王晓冬, 兰亚超, 等. 复合材料学报, 2013, 30(1):135~140. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[4]
章颂云, 宋怀河, 陈晓红. 电源技术, 2002, 26(3):176~179. doi: 10.3969/j.issn.1002-087X.2002.03.014
-
[5]
Wang Y, Su F, Lee J Y, et al. Chem. Mater., 2006, 18(5):1347~1353. doi: 10.1021/cm052219o
-
[6]
陈宁, 王博, 刘洋, 等. 科学通报, 2018, 63(33):3484~3493. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[7]
Zhu X H, Ning C, Fang L, et al. Chin. Sci. Bull., 2011, 56(30):3229~3232. doi: 10.1007/s11434-011-4705-7
-
[8]
Ying S M, Dompablo A D. Energy Environ. Sci., 2009, 2(6):589~609. doi: 10.1039/b901825e
-
[9]
Adams S, Rao R P. J. Mater. Chem., 2012, 22(4):1426~1434. doi: 10.1039/C1JM14588F
-
[10]
Adams S, Rao R P. J. Mater. Chem., 2012, 22(16):7687~7691. doi: 10.1039/c2jm16688g
-
[11]
Thangadurai V, Adams S, Weppner W. Chem. Mater., 2004, 16(16):2998~3006. doi: 10.1021/cm031176d
-
[12]
Adams S, Swenson J. Solid State Ionics, 2002, 154:151~159. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[13]
Adams S. J. Power Sources, 2006, 159(1):200~204. doi: 10.1016/j.jpowsour.2006.04.085
-
[14]
Xiao R, Hong L, Chen L. J. Materiomics, 2015, 1(4):325~332. doi: 10.1016/j.jmat.2015.08.001
-
[15]
Xiao R, Li H, Chen L. Sci. Rep., 2015, 5:14227. doi: 10.1038/srep14227
-
[16]
Zhu J, Lu L, Zeng K. ACS Nano, 2013, 7(2):1666~1675. doi: 10.1021/nn305648j
-
[17]
Yang S, Yan B, Wu J, et al. ACS Appl. Mater. Interf., 2017, 9(16):13999~14005. doi: 10.1021/acsami.6b16321
-
[18]
Zeier W G, Zhou S, Lopez-Bermudez B, et al.ACS Appl. Mater. Interf., 2014, 6(14):10900~10907. doi: 10.1021/am4060194
-
[19]
Liao L, Zuo P, Ma Y, et al. Electrochim. Acta, 2012, 60:269~273. doi: 10.1016/j.electacta.2011.11.041
-
[20]
Kang K, Ceder G. Phys. Rev. B, 2006, 74(9):094105. doi: 10.1103/PhysRevB.74.094105
-
[21]
Lei F, Wei S, Li S, et al. Adv. Energy Mater., 2018, 8(11):1702657. doi: 10.1002/aenm.201702657
-
[22]
Asano T, Sakai A, Ouchi S, et al. Adv. Mater., 2018, 30(44):1803075. doi: 10.1002/adma.201803075
-
[23]
Dai J, Yang C, Wang C, et al. Adv. Mater., 2018, 30(48):1802068. doi: 10.1002/adma.201802068
-
[24]
Kraft M A, Ohno S, Zinkevich T, et al. J. Am. Chem. Soc., 2018, 140(47):16330~16339. doi: 10.1021/jacs.8b10282
-
[25]
http://www.paulingfile.com/
-
[26]
Nosengo N. Nature, 2016, 533(7601):22~25. doi: 10.1038/533022a
-
[27]
Raccuglia P, Elbert K C, Adler P D F, et al. Nature, 2016, 533(7601):73. doi: 10.1038/nature17439
-
[28]
汪洪, 项晓东, 张澜庭. 科技导报, 2018, 36(14):15~21. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[29]
Jain A, Ong S P, Hautier G, et al. Apl Mater., 2013, 1(1):011002. doi: 10.1063/1.4812323
-
[30]
Ceder G. MRS Bull., 2010, 35(9):693~701. doi: 10.1557/mrs2010.681
-
[31]
https://www.mgedata.cn/
-
[32]
Clark S J, Segall M D, Pickard C J, et al. Z. Krist-Cryst Mater., 2005, 220(5/6):567~570. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
-
[33]
Perdew J P, Burke K, Ernzerhof M. Phys. Rev. Lett., 1996, 77(18):3865~3868. doi: 10.1103/PhysRevLett.77.3865
-
[34]
Zhao B, Chen N. Physica C, 2016, 523:1~4. doi: 10.1016/j.physc.2016.02.001
-
[35]
Xiao R, Li H, Chen L. Sci. Rep., 2015, 5:14227. doi: 10.1038/srep14227
-
[36]
Kutteh R, Avdeev M. J. Phys. Chem. C, 2014, 118(21):11203~11214. doi: 10.1021/jp5004402
-
[37]
Jalem R, Nakayama M, Kasuga T. J. Mater. Chem. A, 2014, 2(3):720~734. doi: 10.1039/C3TA13235H
-
[38]
Nakayama M, Kaneko M, Wakihara M. Phys. Chem. Chem. Phys., 2012, 14(40):13963~13970. doi: 10.1039/c2cp42154b
-
[39]
Nakayama M, Jalem R, Kasuga T. Solid State Ionics, 2014, 262:74~76. doi: 10.1016/j.ssi.2013.10.033
-
[40]
Bhattacharya J, van der Ven A. Phys. Rev. B, 2010, 81(10):104304. doi: 10.1103/PhysRevB.81.104304
-
[41]
Ziebarth B, Klinsmann M, Eckl T, et al. Phys. Rev. B, 2014, 89(17):174301. doi: 10.1103/PhysRevB.89.174301
-
[42]
van der Ven A, Ceder G, Asta M, et al. Phys. Rev. B, 2001, 64(18):184307. doi: 10.1103/PhysRevB.64.184307
-
[43]
Lyu Y, Ben L, Sun Y, et al. J. Power Sources, 2015, 273:1218~1225. doi: 10.1016/j.jpowsour.2014.09.082
-
[44]
Xiao R, Li H, Chen L. Chem. Mater., 2012, 24(21):4242~4251. doi: 10.1021/cm3027219
-
[45]
Chen Y, Huo M, Song L, et al. RSC Adv., 2014, 4(80):42462~42466. doi: 10.1039/C4RA07793H
-
[46]
Van Der Ven A, Thomas J C, Xu Q, et al. Phys. Rev. B, 2008, 78(10):104306. doi: 10.1103/PhysRevB.78.104306
-
[47]
Eames C, Clark J M, Rousse G, et al. Chem. Mater., 2014, 26(12):3672~3678. doi: 10.1021/cm5008203
-
[48]
Seo D H, Park Y U, Kim S W, et al. Phys. Rev. B, 2011, 83(20):205127. doi: 10.1103/PhysRevB.83.205127
-
[49]
Islam M M, Bredow T, Heitjans P. J. Phys. Chem. C, 2011, 115(25):12343~12349. doi: 10.1021/jp203045f
-
[50]
Shi S, Qi Y, Li H, et al. J. Phys. Chem. C, 2013, 117(17):8579~8593. doi: 10.1021/jp310591u
-
[51]
Zhang Y, Zhao Y, Chen C. Phys. Rev. B, 2013, 87(13):134303. doi: 10.1103/PhysRevB.87.134303
-
[52]
Du Y A, Holzwarth N. Phys. Rev. B, 2007, 76(17):174302. doi: 10.1103/PhysRevB.76.174302
-
[53]
Lepley N, Holzwarth N, Du Y A. Phys. Rev. B, 2013, 88(10):104103. doi: 10.1103/PhysRevB.88.104103
-
[54]
Xiong K, Longo R, Santosh K, et al. Comput. Mater. Sci., 2014, 90:44~49. doi: 10.1016/j.commatsci.2014.03.030
-
[55]
Clark J M, Eames C, Reynaud M, et al. J. Mater. Chem. A, 2014, 2(20):7446~7453. doi: 10.1039/C3TA15064J
-
[56]
Jalem R, Nakayama M, Kasuga T. Solid State Ionics, 2014, 262:589~592. doi: 10.1016/j.ssi.2013.10.007
-
[57]
Tripathi R, Gardiner G R, Islam M S, et al. Chem. Mater., 2011, 23(8):2278~2284. doi: 10.1021/cm200683n
-
[58]
Kang K, Morgan D, Ceder G. Phys. Rev. B, 2009, 79(1):014305. doi: 10.1103/PhysRevB.79.014305
-
[59]
Islam M M, Bredow T, Minot C. J. Phys. Chem. B, 2006, 110(19):9413~9420. doi: 10.1021/jp0566764
-
[60]
Chen Y, Ouyang C, Song L, et al. J. Phys. Chem. C, 2011, 115(14):7044~7049. doi: 10.1021/jp112202s
-
[61]
Goodenough J B. Phys. Rev., 1955, 100(2):564. doi: 10.1103/PhysRev.100.564

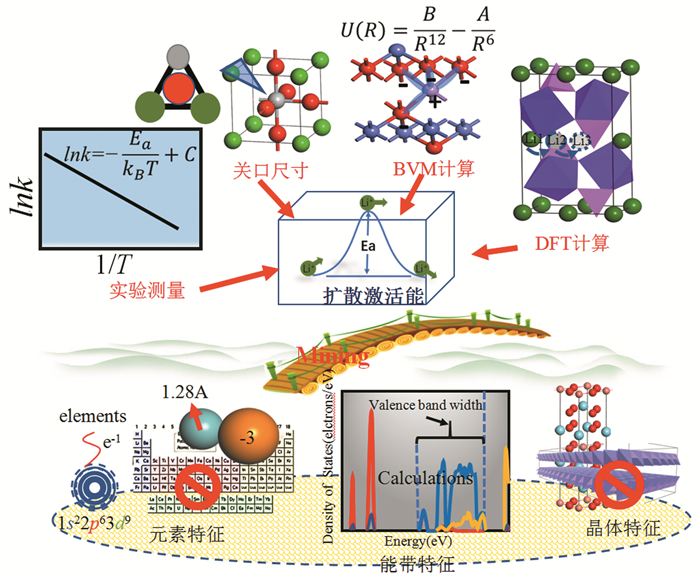
 下载:
下载:
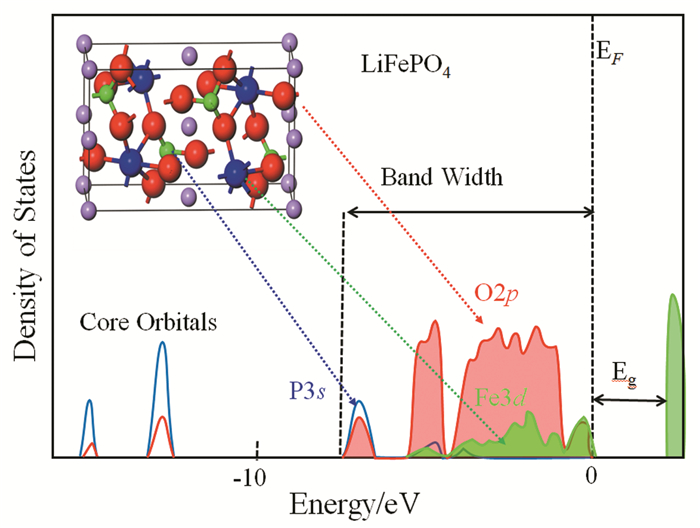
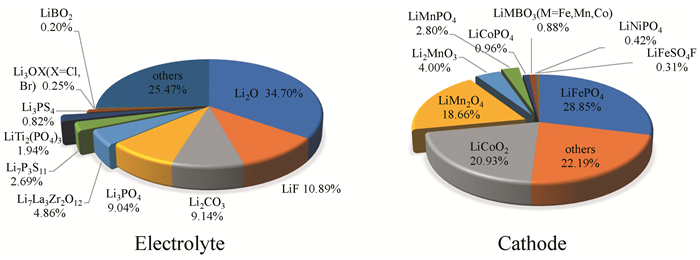
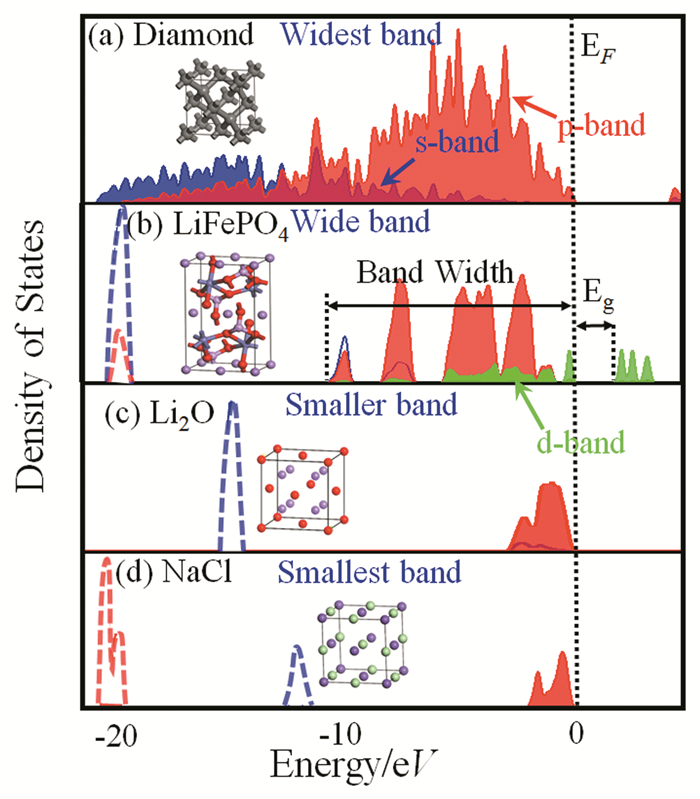

 下载:
下载:








