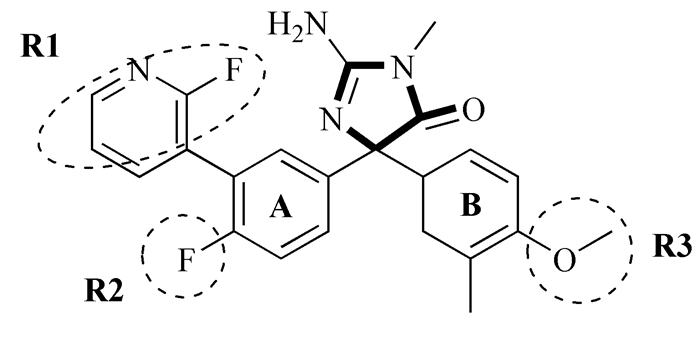
图式 1.
包含共同骨架结构(粗体)的化合物33
Scheme 1.
Molecular 33 containing the common substructure (bold)

阿尔茨海默症(Alzheimer’s disease,AD)作为一种进行性的神经退行性大脑疾病,影响着全世界数以万计的中老年人,带来了严重的社会和经济负担[1, 2]。研究表明,β-淀粉样多肽(Aβ)在大脑中的过量沉积是导致AD的关键因素,抑制Aβ在大脑中的形成和沉积是缓解和治疗AD的有效途径[3~5]。BACE1(β-site APP cleaving enzyme)抑制剂作为修复和治疗AD的潜在手段,已成为目前医学界的重要研究方向[6]。近二十年来已有大量关于BACE1抑制剂的报道,其中一些化合物显示出了良好的脑渗透性,并且在动物模型中显示出低剂量的脑Aβ水平[7]。然而,很少有研究表明BACE1抑制剂能够逆转或减轻AD转基因小鼠模型的记忆和行为缺陷[7, 8]。近年来,已陆续有抑制剂进入3期临床试验,但均因无效而终止试验[9~11],可见新药的开发还是一个耗时耗资的漫长过程[6]。计算机辅助药物设计为新药的研发开辟了新的天地,能够有效节省原料和时间,提高研发效率,目前已广泛应用于临床药物的研发中[12]。
最近,氨基乙内酰脲类化合物作为潜在的BACE1抑制剂,已被多个课题组报道,该类化合物不仅显示出了较强的BACE1抑制活性,且药代动力学特征明显改善。本文采用计算机模拟手段,通过建立实用性较强的构效关系预测模型,从化合物结构上深度挖掘影响分子生物活性的关键因素,揭示其与BACE1的作用特征,为后续新型高效抑制剂的设计和结构改造提供参考[6, 12]。
本实验所研究的105个氨基乙内酰脲类BACE1抑制剂来自于文献[13, 14],其骨架结构如图式 1所示,该类化合物的结构多样性主要体现在R1、R2、R3取代基的不同上。分子的生物活性(以IC50 (μmol/L)表示)均采用荧光共振能量转移酶切割底物的荧光吸收强度测定,模拟实验中用pIC50表示(pIC50=-lg(IC50))。用于模型构建的训练集分子由随机选取的80个化合物组成,剩余分子作为验证集用于模型的外部检验。
本文预测模型的构建采用SYBYL 6.9分子模拟软件程序(TRIPOS公司,St. Louis, MO)。程序中涉及的参数一般均采用缺省值,特别说明的参数除外。分子活性构象的确定是模型建立的首要环节,主要依靠软件中的sketch molecule和minimize模块经分子动力学优化完成。本文选择Tripos力场,加入Gasteiger-Huckel电荷,以能量收敛梯度为0.05kcal·mol-1·Å-1的Powell共轭能量梯度法进行能量最小化运算[6, 12]。最终以最低能量对应的构象作为最佳构象。同时,分子叠合以最强活性分子100为模板,选择图式 1中的加粗部分作为共同骨架进行分子叠合[6, 12]。
CoMSIA作为建立3D-QSAR模型应用较多的方法,采用与距离相关的高斯函数定义立体场(S)、静电场(E)、疏水场(H)、氢键受体场(A)以及氢键供体场(D)的分子场特征。当结构类似的药物以相似的模式作用于同一受体时,药物周围的分子场能量势函数分布决定了其活性的大小,以所得分子场信息作为自变量对药物活性进行回归分析,即可建立定量构效关系模型[6]。CoMSIA法按照2.0Å的步长均匀划分产生格点,每个格点上用sp3杂化的C+作为探针离子来评价分子场特征[6, 12]。本实验将场能阈值设定0.3kcal·mol-1,其他参数为系统缺省值。各分子场的计算公式为:
|
$ A_{F, k}^{q}\left( j \right) = - \sum {\omega _{{\rm{probe, k}}}}{\omega _{{\rm{ik}}}}{e^{ - \alpha r_{{\rm{iq}}}^2}} $ |
式中,k代表各分子场;i代表分子j中的原子序号;ωprobe, k代表探针离子的某种分子场特征(电荷为+1,原子半径为1Å,疏水性为+1,氢键给体和受体强度为+1);ωik是i原子中物理化学性质k的实际值;α为衰减因子(attenuation factor);riq则表示探针离子在某格点q上和分子中i原子之间的距离[6, 12]。
模型的内部检验采用非交叉验证(non-cross validation)法进行,以非交叉验证相关系数Rncv2以及标准偏差SEE为主要评价指标。由25个验证集分子对模型进行外部检验,得到相关系数Rpre2。实验建立的CoMSIA模型具有较好的相关性、较强的稳定性和预测能力。因此,基于训练集分子建立的CoMSIA模型完全可以用于预测具有相似骨架的BACE1抑制剂的生物活性[6, 12]。
分子对接技术可揭示药物分子与蛋白质受体之间的结合模式,两者之间的结合能越低,说明结合越稳定[6, 12]。本文采用GOLD 5.1程序,运用遗传算法对105个氨基乙内酰脲类BACE1抑制剂进行分子对接。本文选取的BACE1蛋白质的晶体结构(PDB代码:3INH)来源于蛋白质数据库Protein Data Bank(www.rcsb.org/pdb),分辨率为1.80Å。
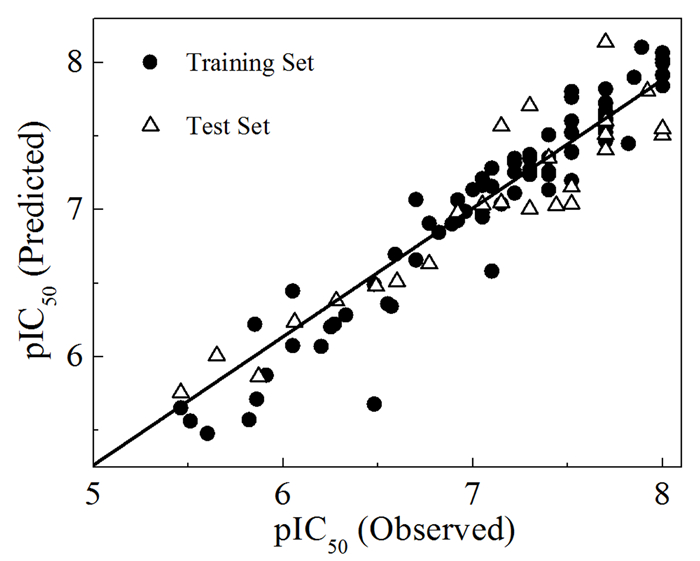
运用留一法(LOO)对CoMSIA中5种分子场的31种组合形式进行回归分析。结果表明,以S、E、H、A场建立的模型最佳,所得交叉验证相关系数Q2=0.45,最佳主成分数OPN=7,非交叉验证相关系数Rncv2=0.87,外部验证相关系数Rpre2=0.85(表 1),意味着模型的预测能力较强,具有显著的统计意义。而且,从分子活性实验值与预测值的线性相关回归曲线(图 1)也可以看出,数据点大都均匀分布在回归线的附近,表明本实验建立的CoMSIA模型具有较好的相关性、较强的稳定性和预测能力。因此,基于训练集分子建立的CoMSIA模型完全可以用于预测具有相似骨架的BACE1抑制剂的生物活性[6, 12]。
 下载:
导出CSV
下载:
导出CSV
| Index | Q2 | OPN | Rncv2 | Rpre2 | SEE | Contributions | |||
| S | E | H | A | ||||||
| CoMSIA | 0.45 | 7 | 0.87 | 0.85 | 0.27 | 0.20 | 0.31 | 0.28 | 0.21 |
CoMSIA模型的三维等势面图可以直观反映各分子场对化合物区域基团的影响,以此得到提升抑制剂活性的特征结构信息,指导化合物的结构改造。4个分子场的等势面图中均有两种不同颜色的云团(图 2),它们分别代表相应分子场对化合物区域基团的不利及有利影响。
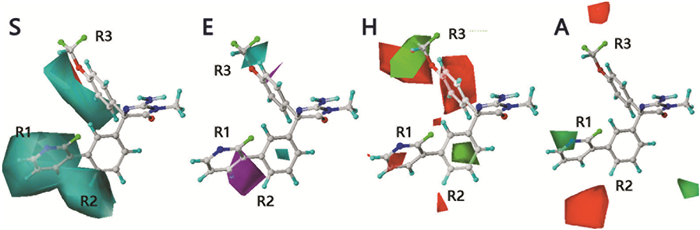
立体场S在R1取代基附近以及与R3相连的苯环上方均出现了青蓝色和红色等势面云图,且均为青蓝色有利云团包覆红色不利云团,说明该区域为受立体场影响的敏感区域。例如,化合物4(pIC50=5.51)和7(pIC50=7.15)在结构上的不同之处在于与R1和R2取代基相连的A环,化合物7链接的是大体积的金刚烷基,化合物4的为苯环,可见,拥有大体积基团的7表现出更强的BACE1抑制活性。然而,具有不同R1取代基的化合物78(pIC50=8.00)和79(pIC50=7.00)却表现出了相反的构效信息,链接大体积烷氧基的化合物79的抑制活性明显低于78,说明R1和R2取代基所处的区域对立体场比较敏感。
静电场E中,R3取代基附近有较大的蓝色云团,表明该位置链接正电基团将有利于抑制活性的增强,例如R3取代基为正电基团(-OCH3)的分子98(pIC50=7.52),其抑制活性强于拥有强吸电子基团OCF3的分子99(pIC50=7.22)。R1取代基附近有一片紫色不利云团,说明该区域负电基团的存在能够增强抑制剂。例如,在结构上,化合物99(pIC50=7.22)和102(pIC50=5.85)唯一区别就是R1取代基的不同,2-F-吡啶基的电负性强于氢原子,因此,化合物99表现出了较高的抑制活性。
在疏水场H中,绿色有利云团和红色不利云团同时出现在R3取代基附近,表明该区域为疏水场的敏感区域。在R3取代基上,化合物8结合了较7疏水性更强的-OCH2CH3,却表现出了较低的抑制活性;化合物103(pIC50=6.77)结合的-OCHF2其疏水性强于化合物102(pIC50=5.85)的-OCF3基团,却表现出了较强的分子活性,验证了该区域为疏水场敏感区域的等势面结论。此外,在R1和R2取代基周围也有小块红色和绿色云团出现,表明该区域对疏水场较敏感。例如,化合物63(pIC50=7.70)、64(pIC50=7.40)、65(pIC50=7.52),它们的R1取代基疏水性依次增强,但是化合物64的抑制活性最低,这与以上结论一致。
在氢键受体场A中,R1取代基附近出现了绿色有利云团以及红色不利云团,说明该区域结合氢键受体或供体基团均有利于增强分子活性。例如,R1取代基分别为氢键受体基团的化合物100和氢键供体基团的化合物74,pIC50均为8.00。此外,在R3取代基附近有一小块红色不利云团,说明此处不利于结合较多的氢键受体基团。在B环4位结合了氢键受体基团的化合物16(pIC50=7.05)、21(pIC50=7.15)、26(pIC50=6.82),其抑制活性均低于结合甲基的化合物15。
分子对接通过寻找合理的取向和构象,将药物配体与蛋白质受体的活性位点进行对接,从而揭示两者之间的结合模式,为后续抑制剂的设计和结构改造进行指导。本实验将最大活性分子33作为模板化合物与BACE1蛋白质进行对接,结果如图 3。对接空腔中的氨基酸残基主要有Asp32、Asp228、Gly34、Thr232、Ser229、Ile110、Thr294、Tyr71、Trp76、Gly230等,与原文献晶体复合物的残基信息一致,说明本实验准确定位了作用空腔[13, 14]。
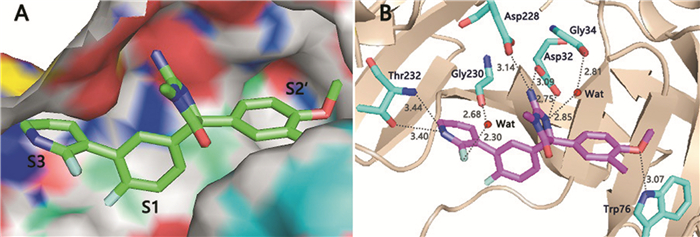
(A)化合物33与BACE1的结合空腔(PDB: 3INH);(B)化合物33与BACE1氨基酸残基的结合作用图
从图 3(A)可以看出,该类抑制剂主要占据了BACE1受体空腔的S3、S1、S2′活性位点。其中,氟代吡啶基(R1取代基)占据了S3活性位点,吡啶氮作为氢键受体,与氨基酸Thr232形成了较强的氢键作用力(—N…HN—,3.44Å;—N…HO—,3.40Å),如图 3(B)所示。同时,水分子作为连接配体与蛋白质的桥梁,可促进二者之间的结合。吡啶环上的氟取代基通过水分子与氨基酸Gly230产生氢键结合(—F…HO—,2.30Å;—O…HO—,2.68Å)。从图 3(A)可以看出,吡啶环占据的S3活性位点空间相对宽阔,R1取代基区域适宜结合大体积的基团,但是,由于多个氢键作用力的形成,使S3位点上方的区域变得狭小,又不利于结合大体积的取代基,因此,该区域为空间立体场S的敏感区域。与大多数BACE1抑制剂的作用模式类似,抑制剂33的氨基乙内酰脲结构作为功能基团与BACE1受体催化活性中心的关键氨基酸Asp32和Asp228产生了较强的氢键结合。其中,氢键供体氨基分别与Asp32、Asp228形成氢键作用力(—HN…O—,3.09Å;—HN…O—,3.14Å);氢键受体基团乙内酰脲的氮与Asp32也存在氢键作用力。此外,乙内酰脲氮作为氢键受体基团通过水分子桥连,与氨基酸Gly34产生氢键结合力(—N…HO—,2.85Å;—OH…O—,2.81Å)。
分子33的甲氧基(R3取代基)占据了受体活性空腔的S2′位点。甲氧基作为氢键受体与氨基酸Trp76产生氢键作用力(—O…HN—,3.07Å)。此外,分子33的A环和R2取代基占据了BACE1靶标的S1活性位点。该位点具有高度疏水性,形状近似球形[13, 14]。若增加配体R1取代基的体积和亲脂性,可以增强配体和蛋白质分子之间的亲和力。如将靠近R1处的取代基置换为金刚烷基,可能会使抑制活性大大增强[13]。
通过与原文献对比分析发现,作用空腔中的Asp32、Asp228和Trp76残基与配体间的氢键作用模式与原文献(配体为化合物15)完全一致[13]。所不同的是原文献中吡啶氮(R1取代基)与Ser229通过水分子桥连产生氢键结合,而本实验中是与Thr232直接形成两个强有力的氢键作用;此外,本实验中催化活性中心的乙内酰脲氮与氨基酸Gly34之间也存在氢键结合力。造成以上差异的原因可能是化合物15与33具有不同的R1和R2取代基(见表 2),产生的空间构象稍有区别。
下载:
导出CSV
 |
||||||
| Compd. | Skeleton | R1 | R2 | X | R3 | pIC50 |
| 4 | A | H | H | H | OMe | 5.51 |
| 7 | B | — | — | H | OMe | 7.15 |
| 8 | B | — | — | H | OEt | 6.48 |
| 15 | A | 3-Pyridyl | H | Me | OMe | 7.40 |
| 16 | A | 3-Pyridyl | H | OMe | OMe | 7.05 |
| 21 | A | 3-Pyridyl | H | O-cyclopentyl | OMe | 7.15 |
| 26 | A | 3-Pyridyl | H | CN | OMe | 6.82 |
| 63 | A | MeC≡C | H | H | OCHF2 | 7.70 |
| 64 | A | MeCH2C≡C | H | H | OCHF2 | 7.40 |
| 65 | A | Me(CH2)2C≡C | H | H | OCHF2 | 7.52 |
| 74 | A | HO(CH2)2C≡C | H | H | OCHF2 | 8.00 |
| 78 | A | F(CH2)3O | F | H | OCHF2 | 8.00 |
| 79 | A | F(CH2)4O | F | H | OCHF2 | 7.00 |
| 98 | A | 2-F-3-Pyridyl | H | H | OMe | 7.52 |
| 99 | A | 2-F-3-Pyridyl | H | H | OCF3 | 7.22 |
| 100 | A | 2-F-3-Pyridyl | H | H | OCHF2 | 8.00 |
| 102 | A | H | H | H | OCF3 | 5.85 |
| 103 | A | H | H | H | OCHF2 | 6.77 |
Tg2576小鼠的体内药效学实验结果表明,以100mg/kg po急性给药抑制剂33,8h后测得血浆中Aβ40显著降低69%(p < 0.001)[15],类药特性明显。因此,进一步挖掘其结构特征以及与蛋白受体间的结合模式,对于增强配体-BACE1酶作用位点之间的亲和力、改善配体的脑中心暴露度具有重要意义。
在通常的药物模拟计算中,水分子作为溶剂在对接之前往往会被删除[16]。研究表明,在溶剂化和去溶剂化过程中,水分子在配体-蛋白质之间的结合过程中发挥着非常重要的桥梁作用[17]。而且,BACE1催化裂解APP的过程中,水分子的存在有助于APP的Met和Asp间肽键的催化裂解[18]。本文对接结果也表明(图 3B),水分子在配体与氨基酸残基Gly230、Gly34的结合中起到了关键的桥梁作用,促进了化合物抑制活性的提升。
本文以105个氨基乙内酰脲类BACE1抑制剂为研究对象,建立了预测能力较强的CoMSIA模型(Q2=0.45,Rncv2=0.87,Rpre2=0.85),对分子生物活性与结构之间的构效关系进行了定量研究,全面考察了配体与受体的结合方式,揭示了配体与受体蛋白质间的作用机制:(1)氨基乙内酰脲取代基作为核心功能基团,同BACE1催化活性中心的Asp32和Asp228形成了较强的氢键作用力,增强了抑制剂分子的抑制活性;(2)占据S3和S2′活性位点的R1和R3取代基分别与受体氨基酸产生了较强的氢键结合;(3)R1和R2取代基所在区域为立体场的敏感区域。由立体场、静电场、疏水场以及氢键受体场共同建立的CoMSIA模型预测能力较强,由CoMSIA等势面图分析以及分子对接等方法得出影响氨基乙内酰脲类BACE1抑制剂活性的特征结构。实验所得上述信息为后续抑制剂的结构优化提供了重要参考。
Burns A, Iliffe S. Brit. Med. J., 2009, 338: b158.
Berchtold N C, Cotman C W. Neurobiol. Aging, 1998, 19(3): 173~189.
Hardy J, Allsop D. Trends Pharmacol. Sci., 1991, 12(10): 383~388.
Mudher A, Lovestone S. Trends Neurosci., 2002, 25(1): 22~26.
Pimplikar S W. Int. J. Biochem. Cell Biol., 2009, 41(6): 1261~1268.
吴倩, 高庆平, 李丹, 等.中国生物化学与分子生物学报, 2018, 34(7): 740~746.
Coimbra J R M, Marques D F F, Baptista S J, et al. Front. Chem., 2018, 6: 178.
Imbimbo B P, Watling M. Expert. Opin. Investig. Drugs, 2019, 28(11): 967~975.
Strobel G. Alzheimer Res. Forum, 2019, 17.
Lo A C, Duggan Evans C, Mancini M, et al. J. Prev. Alzheimers Dis., 2018, 5(Suppl 1): S37.
Shugart J, Strobel G. Alzheimer Res. Forum, 2019, 12.
吴倩, 高庆平, 刘莉, 等.化学通报, 2018, 81(8): 745~752.
Malamas M S, Erdei J, Gunawan I, et al. J. Med. Chem., 2010, 53: 1146~1158.
Malamas M S, Robichaud A, Erdei J, et al. J. Bioorg. Med. Chem. Lett., 2010, 20: 6597~6605.
Hsiao K, Chapman P, Nilsen S, et al. Science, 1996, 274: 99~102.
Van Der Spoel D, Lindahl E, Hess B, et al. J. Comput. Chem., 2005, 26: 1701~1718.
Aghaee E, Ghasemi J B, Manouchehri F, et al. J. Mol. Model., 2014, 20: 1~13.
Yuan J, Venkatraman S, Zheng Y, et al. J. Med. Chem., 2013, 56: 4156~4180.

图式 1 包含共同骨架结构(粗体)的化合物33
Scheme 1 Molecular 33 containing the common substructure (bold)

图 1 抑制剂生物活性实验值与预测值的线性相关图
Figure 1 Plots of the actual versus the predicted pIC50 values for the CoMSIA model

图 2 基于BACE1抑制剂分子100的COMSIA等势面图(S:立体场;E:静电场;H:疏水场;A:氢键受体场)
Figure 2 CoMSIA contour maps superimposed on compound 100

图 3 分子对接图
Figure 3 The docking results
(A)化合物33与BACE1的结合空腔(PDB: 3INH);(B)化合物33与BACE1氨基酸残基的结合作用图
表 1 COMSIA所得PLS分析结果及各分子场对模型的贡献率
Table 1. PLS statistics of CoMSIA model
| Index | Q2 | OPN | Rncv2 | Rpre2 | SEE | Contributions | |||
| S | E | H | A | ||||||
| CoMSIA | 0.45 | 7 | 0.87 | 0.85 | 0.27 | 0.20 | 0.31 | 0.28 | 0.21 |
 下载: 导出CSV
下载: 导出CSV
表 2 氨基乙内酰脲类BACE1抑制剂的结构和生物活性
Table 2. Structure-activity relationship data related to aminohydantoins as BACE1 inhibitors
|
||||||
| Compd. | Skeleton | R1 | R2 | X | R3 | pIC50 |
| 4 | A | H | H | H | OMe | 5.51 |
| 7 | B | — | — | H | OMe | 7.15 |
| 8 | B | — | — | H | OEt | 6.48 |
| 15 | A | 3-Pyridyl | H | Me | OMe | 7.40 |
| 16 | A | 3-Pyridyl | H | OMe | OMe | 7.05 |
| 21 | A | 3-Pyridyl | H | O-cyclopentyl | OMe | 7.15 |
| 26 | A | 3-Pyridyl | H | CN | OMe | 6.82 |
| 63 | A | MeC≡C | H | H | OCHF2 | 7.70 |
| 64 | A | MeCH2C≡C | H | H | OCHF2 | 7.40 |
| 65 | A | Me(CH2)2C≡C | H | H | OCHF2 | 7.52 |
| 74 | A | HO(CH2)2C≡C | H | H | OCHF2 | 8.00 |
| 78 | A | F(CH2)3O | F | H | OCHF2 | 8.00 |
| 79 | A | F(CH2)4O | F | H | OCHF2 | 7.00 |
| 98 | A | 2-F-3-Pyridyl | H | H | OMe | 7.52 |
| 99 | A | 2-F-3-Pyridyl | H | H | OCF3 | 7.22 |
| 100 | A | 2-F-3-Pyridyl | H | H | OCHF2 | 8.00 |
| 102 | A | H | H | H | OCF3 | 5.85 |
| 103 | A | H | H | H | OCHF2 | 6.77 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们