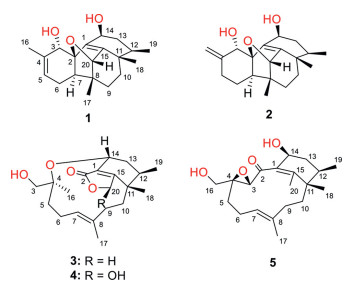
Figure 1.
Chemical structures of compounds 1‒5.
Hawanoids A‒E, unprecedented diterpenoids with PAF-induced platelet aggregation inhibitory activities from the deep-sea-derived fungus Paraconiothyrium hawaiiense
Shushuai Chen , Hongxin Liu , Saini Li , Yuchan Chen , Wei Ye , Haohua Li , Haibo Tan , Dongli Li , Zhaoming Liu , Weimin Zhang

Phomactin family is a type of unusual diterpenoids featuring a cyclohexenyl ring with a macrocyclic "strap" that bridges it [1]. Due to their fascinating bicyclo[9.3.1]pentadecane framework and PAF (platelet activating factor) antagonistic activities, phomactins have inspired great interest of natural product researchers and synthetic chemists for almost 30 years [1-4]. Since the first phomactin derivative (phomactin A) was isolated from a crab shell derived fungus Phoma sp. [5], only 24 analogues have been discovered from three different fungal species to date, including phomactins A‒G, phomacta-1(14),3,7-triene and Sch 49027 from Phoma sp. [6-8], phomactins H‒P from an unidentified fungus MPUC046 [9-12], and phomactins Q‒V from Biatriospora sp. [1, 13] Though all of them shared a common bicyclic skeleton, the oxidation and cyclization always led to a changeable functionality, for example the phomactin A which constructed a tetracyclic framework is the most complex member of phomactin family to date. Given the potential biological value and undiscovered structure diversity, it is expected and worth exploring novel phomactin family members with unprecedented skeletons.
Deep-sea derived fungi, which were collected from the sediment or water at a depth over 1000 m, have attracted considerable attention because of their extreme living environment as well as their potential abilities to produce structurally novel and bioactive natural products [14-17]. Our group has endeavoured to discover novel bioactive molecules from deep-sea-derived fungi for over ten years. Recently, the chemical investigation of the marine-derived fungus Paraconiothyrium hawaiiense FS482 collected from the sediment of Indian Ocean led to the isolation of five highly cyclized phomactin familial diterpenoids, hawanoids A‒E (1‒5) (Fig. 1). Among them, compounds 1 and 2, constructing a complicated tetracyclo[6.6.2.02,7.011,15]cetane carbon skeleton, represented a new type of highly cyclized framework belonging to phomactin family. Meanwhile, 3 and 4 were found to be the first examples of ring-opening oxidative rearranged analogues of phomactin family by possessing an unusual 11, 14-macrocyclic ether moiety. Herein, the details of the isolation, structure identification and biological assay of the new compounds were discussed.
Hawanoid A (1) was obtained as a colorless block crystal. The molecular formula was deduced to be C20H28O3 based on the sodium adduct HRESIMS peak at m/z 337.1770 [M + Na]+ (calcd. 337.1774, C20H28O3Na). The 1H NMR spectrum (Table S1 in Supporting information) displayed the signals of four methyl groups, three oxygenated methine groups, an olefin proton and a series of shielded aliphatic protons. Further analysis of 13C NMR and HSQC spectra revealed the presence of 20 carbons, which could be divided into four methyl groups, four methylenes, six methines and six quaternary carbons (including a full-substituted double bond). Considering of the degrees of unsaturation, hawanoid A (1) should be a penta-cyclic diterpenoid.
COSY cross-peaks (Fig. 2) of H-5/H-6/H-7, H2–9/H2–10 and H3–19/H-12/H2–13/H-14 resolved the three independent coupling fragments in 1. The HMBC correlations (Fig. S5 in Supporting information) of H3–16 to C-3/C-4/C-5 as well as H-3/H-7/H2–6 to C-2 constructed ring A. Ring B could be deduced by the correlations from H3–17 to C-20/C-8/C-9, H3–18 to C-10/C-11/C-15 and H-20 to C-1, while ring C could be evaluated on the basis of HMBC signals from H-14 to C-1/C-15 and from H3–19 to C-11. The further key correlation between H3–17 and C-7 suggested that ring A and ring B were connected through C-7 and C-8. Moreover, the cross-peak between H-7 and C-1 could be detected in HMBC spectrum, which indicated that ring A and ring C were closed via C-1‒C-2 to form ring D. At this point, a tetracyclo[6.6.2.02,7.011,15]cetane carbon skeleton was established. Finally, an oxygen bridge from C-2 to C-20 should be constructed according to the deshielded chemical shifts and the degrees of unsaturation. Hence, compound 1 was elucidated to be a multi-cyclic diterpenoid, confirmed by the X-ray crystallographic analysis (Fig. 2).
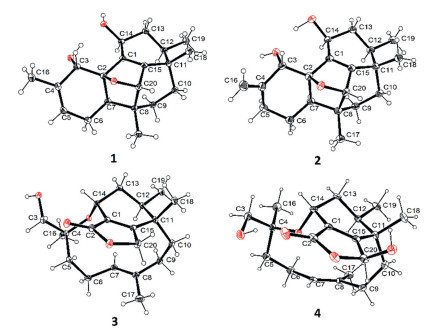
The relative configuration could be deduced by NOE correlations (Fig. S1 in Supporting information) and confirmed by X-ray diffraction. The correlations of H-7/H2–9/H-12/H-14 indicated that they were co-oriented and revealed a cage-shaped structural feature of ring B, C and D. The deficiency of the cross-peak between H-7 and H-3 suggested the anti-orientation of them. The Flack parameter of ‒0.03(10) suggested the absolute configuration should be consistent with the obtained result. Furthermore, a quantum chemical calculation of the ECD spectra at b3lyp/6–311+g(d, p) level was also illustrated to confirm the point. The results exhibited an excellent fit between the calculated plot and the experimental spectrum of 1, which gave a positive Cotton Effect at 215 nm (Fig. S6 in Supporting information). Thus, the absolute configuration was assigned as 2R, 3S, 7R, 8R, 11S, 12R, 14S and 20S unambiguously.
Hawanoid B (2) gave the same molecular formula as that of compound 1 according to the deprotonated HRESIMS peak at m/z 315.1945 [M ‒ H]‒. The NMR spectra (Table S1) recorded in methanol were similar to those of 1, except for the absence of the Me-16 (δH 1.84) and the presence of an additional methylene at δH 2.45/2.30. Moreover, a terminal alkenyl moiety could be concluded through the characteristic signals of δH 5.05/4.92 (each brs) and δC 110.3/148.1 (C-16/C-4). All the above evidences suggested that the tri-substituted double bond Δ4 in 1 has been isomerized to a di-substituted one in 2 (Δ4,16). Comprehensive analysis of 2D NMR data and X-ray diffraction confirmed the gross structure as shown (Fig. S5 and Fig. 2). Since the shielded signals in 1H NMR were overlapped, it was hard to evaluated the relative configuration by NOE correlations. Thus, the relative configuration was directly elucidated from X-ray analysis. The similar ECD spectrum (Fig. S7 in Supporting information) exhibiting a positive Cotton effect at about 230 nm as well as the Flack parameter of 0.11 (11) indicated the same absolute configuration as that of 2.
Hawanoid C (3), isolated as a colorless crystal, showed the molecular formula of C20H30O4, which was evaluated by the protonated ion peak at m/z 335.2223 [M + H]+ from HRESIMS. The IR spectrum indicated that an α,β-unsaturated ester carbonyl moiety (1716 cm‒1). The 1H and 13C NMR spectra (Table S2 in Supporting information) combined with the HSQC spectra resolved 20 carbon signals composed of four methyl groups, seven methylenes, three methines and six quaternary carbons (including an ester carbonyl at δC 176.7 and a full-substituted double bond). Comprehensive analysis of the 1D NMR spectra between 3 and 1/2 suggested that hawanoid C (3) was also a diterpenoid with a different cyclic framework.
The complete structure was determined by the further analysis of the 2D NMR data. 1H-1H-COSY cross peaks of H3–19/H-12/H-13/H-14 as well as the HMBC correlations from H3–18 to C-15/C-11/C-12 and H-14 to C-1/C-15 constructed the similar ring C as those in 1 and 2. Considering of deshielded chemical shift of C-15 (δC 177.2) and the shielded shift of H2–20 (δH 4.96 and 4.86), an α,β-unsaturated lactone moiety (ring B) should be existed, evidenced by the correlations from H2–20 to C-1 and C-15. Moreover, a C8 aliphatic chain (C-3 ~ C-10) was concluded based on the COSY cross peaks of H-5/H-6/H-7 and H-9/H-10 as well as the HMBC correlations from H3–16 to C-3/C-4/C-5 and from H3–17 to C-7/C-8/C-9. Finally, according to the key correlations from H-14 and H3–18 to C-4 and C-10, respectively, a macrocyclic ether (ring A) could be evaluated between the C8 aliphatic chain and ring C, which was further confirmed by the deshielded chemical shift of H-14 (δH 4.34) and the degrees of hydrogen deficiency. Hence, compound 3 was established to be tricyclic diterpenoid possessing an unusual 11, 14-macrocyclic ether structural feature, which belongs to an oxidative rearranged member of phomactin family. The relative configuration was determined through NOESY spectrum (Fig. S1) and X-ray crystallization analysis while the absolute configuration was deduced to be 4S, 11S, 12R by comparing the theoretical ECD spectrum at b3lyp/6–311+g(d, p) level with the experimental plot (Fig. S8 in Supporting information).
Hawanoid D (4) exhibited the similar 1H and 13C NMR data (Table S2) with those of 3, suggesting they were homologues. The IR spectrum also exhibited an α,β-unsaturated ester absorption at 1720 cm.‒1 The most obvious difference was that one of the oxygen-bearing methylene (δH 4.96/4.86; δC 71.9) has been converted to a hemiacetal moiety (δH 6.23; δC 100.5). The deprotonated ion peak at m/z 349.2023 [M ‒ H]‒ in HRESIMS spectrum further supported the point. By resolving the 2D NMR spectra, the planar structure of 4 could be constructed as shown.
The relative configuration was elucidated on the basis of the NOESY correlations and the H—H coupling constants (Fig. S1), which were similar to those of compound 3. The X-ray diffraction analysis further confirmed the established relative configuration and the similar experimental ECD spectra of 3 and 4 suggested that they shared the same absolute configuration (Fig. S8).
Hawanoid E (5) was obtained as a colorless powder, giving a molecular formula of C20H30O4 based on the sodium adduct peak of m/z 357.2036 from HRESIMS. The absorption at 1689 cm‒1 in IR spectrum suggested that a α,β-unsaturated ketone instead of ester carbonyl. By comparing its NMR data (Table S3 in Supporting information) with that of the known diterpenoid phomactin B1 isolated from a marine-derived fungus Phoma sp. [5], it could be elucidated that they shared the same diterpenoid framework. The major differences were the absence of the olefin proton (H-14 in phomactin B1) and the presence of an additional hydroxymethyl group (H2–16 in 5). Further analysis of the 2D NMR spectroscopic data led to the establishment of the planar structure of 5, which was an isomer of phomactin B1. The further NOESY analysis (Fig. S1) and ECD (Fig. S9 in Supporting information) calculations enabled us to determine the absolute configuration as 3S, 4R, 11S, 12R and 14S.
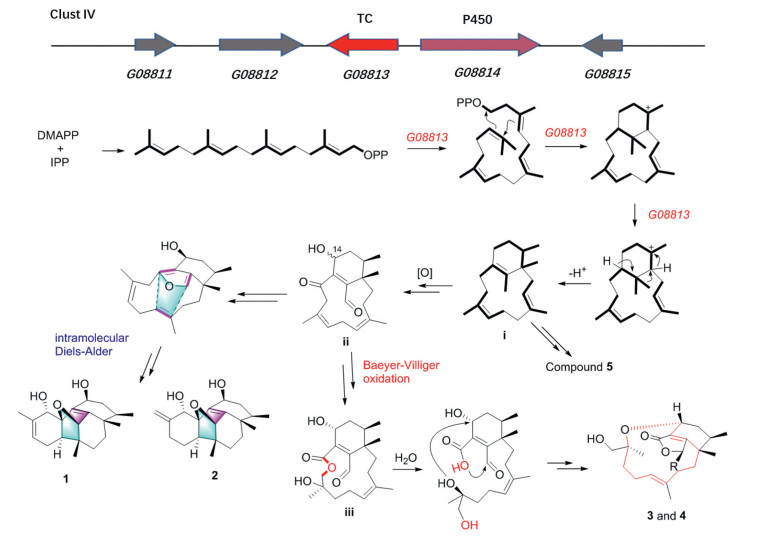
To be best of our knowledge, all of the reported phomactins constructed the same bicyclo[9.3.1]pentadecane framework with different post-modification to date. This is the first example of phomactins exhibited unprecedented skeletons. The hypothetical biosynthetic pathway (Fig. 3) involving intramolecular Diels-Alder reaction and Baeyer-Villiger oxidation was proposed based on the speculated biosynthetic gene cluster Ⅳ (Figs. S2-S4 in Supporting information).
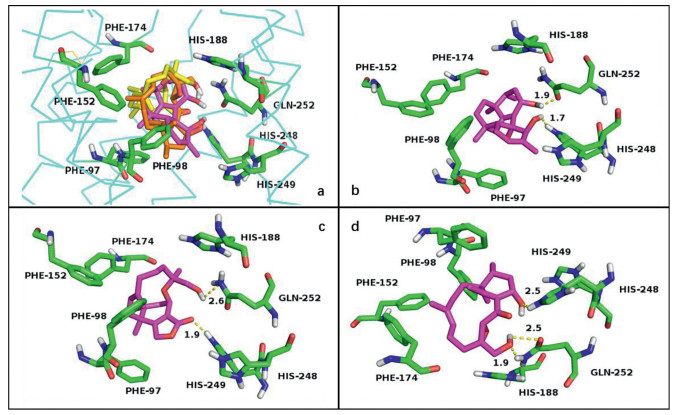
Hawanoids A‒E were evaluated for their inhibitory activities against PAF-induced platelet aggregation. The results indicated that compounds 3 and 5 exhibited significant activities with the IC50 values of 7.1 and 8.9 µmol/L while compounds 1, 2 and 4 showed moderate activities at 50 µmol/L with the IC50 values in the range of 15‒67 µmol/L (Table S5 in Supporting information). Furthermore, in order to understand the interaction between the PAF receptor and hawanoids with different skeletons, the molecular docking study was carried out (Fig. 4). The human PAF receptor protein (PDBID: 5ZKP) was used as macromolecule and compounds 1, 3, 5 which represented three types of skeleton were set to be ligands. The results indicated that the common hydrophobic units from C-5 to C-12 in compounds 1, 3 and 5 help them bind into the hydrophobic pocket of protein composing of Phe97, Phe98, Phe152, Phe174 with a similar binding pose. Besides, the 14-OH/3-OH in 1, 3-OH/C=O at C-2 in 3 as well as the 16-OH/14-OH in 5 constructed a "dentate" in space, which could form hydrogen bonds to the key active residue His252 and His248 [18], respectively.
In conclusion, five novel diterpenoids belong to phomactin family were isolated from the deep-sea-derived fungus P. hawaiiense FS482. This is the first report of phomactins possessed an unprecedented tetracyclo[6.6.2.02,7.011,15]cetane carbon skeleton (1 and 2) and an unusual 11, 14-macrocyclic ether moiety (3 and 4), which will make contributions to the chemical and biological diversities of diterpenoids from deep-sea fungal resources.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was funded by National Natural Science Foundation of China (Nos. 41906106, 31272087), the Natural Science Foundation of Guangdong Province, China (No. 2021A1515011416), Guangdong Special Support Program (No. 2019TQ05Y375) and Guangdong Provincial Special Fund for Marine Economic Development Project (No. GDNRC [2021]054).
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Kuroda, K.J. Nicacio, I.A. Silva-Jr, et al., Nat. Chem. 10 (2018) 938–945. doi: 10.1038/s41557-018-0084-x
J. Ciesielski, A. Frontier, Org. Prep. Proced. Int. 46 (2014) 214–251. doi: 10.1080/00304948.2014.903142
J. Huang, W. Bao, S. Huang, et al., Org. Lett. 20 (2018) 7466–7469. doi: 10.1021/acs.orglett.8b03242
R. Guo, H. Zhai, Li Y, Chin. Chem. Lett. 32 (2021) 1400–1402. doi: 10.1016/j.cclet.2020.09.023
M. Sugano, A. Sato, Y. Iijima, et al., J. Am. Chem. Soc. 113 (1991) 5463–5464. doi: 10.1021/ja00014a053
M. Sugano, A. Sato, Y. Iijima, et al., J. Org. Chem. 59 (1994) 564–569. doi: 10.1021/jo00082a012
M. Sugano, A. Sato, Y. Iijima, et al., J. Antibiot. 48 (1995) 1188–1190. doi: 10.7164/antibiotics.48.1188
M. Chu, I. Truumees, I. Gunnarsson, et al., J. Antibiot. 46 (1993) 554–563. doi: 10.7164/antibiotics.46.554
K. Koyama, M. Ishino, K. Takatori, et al., Tetrahedron Lett. 45 (2004) 6947–6948. doi: 10.1016/j.tetlet.2004.07.071
M. Ishino, N. Kiyomichi, K. Takatori, et al., Tetrahedron 66 (2010) 2594–2597. doi: 10.1016/j.tet.2010.02.040
M. Ishino, K. Kinoshita, K. Takahashi, et al., Tetrahedron 68 (2012) 8572–8576. doi: 10.1016/j.tet.2012.08.013
M. Ishino, H. Kamauchi, K. Takatori, et al., Tetrahedron Lett. 57 (2016) 4341–4344. doi: 10.1016/j.tetlet.2016.08.016
M.R.Z. Passarini, C. Santos, N. Lima, R.G.S. Berlinck, L.D. Sette, Arch. Microbiol. 195 (2013) 99–111. doi: 10.1007/s00203-012-0854-6
G. Daletos, W. Ebrahim, E. Ancheeva, et al., Curr. Med. Chem. 22 (2018) 186–207.
Y.T. Wang, Y.R. Xue, C.H. Liu, Mar. Drugs 13 (2015) 4594–4616. doi: 10.3390/md13084594
C.L. Xie, D. Zhang, K.Q. Guo, et al., Chin. Chem. Lett. 33 (2022) 2057–2059. doi: 10.1016/j.cclet.2021.09.073
J. Xu, H. Tan, Y. Chen, et al., Chin. Chem. Lett. 30 (2019) 439–442. doi: 10.1016/j.cclet.2018.09.018
C. Gui, W. Zhu, G. Chen, et al., Proteins 67 (2007) 41–52. doi: 10.1002/prot.21213

Figure 3 The cluster Ⅳ in the genome of P. hawaiiense FS482 responsible for the hawanoids biosynthesis and the possible biosynthetic pathway of 1‒5.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

