引用本文:
王珏, 杨勇, 青明, 白云坡, 王洪, 胡彩霞, 相宏伟, 岳仁亮. 助剂对铁基费托合成催化剂氧化行为的影响:H2O作用的解读[J]. 燃料化学学报,
2020, 48(1): 63-74.
Citation:
WANG Jue, YANG Yong, QING Ming, BAI Yun-po, WANG Hong, HU Cai-xia, XIANG Hong-wei, YUE Ren-liang. Effect of the promoters on oxidation behavior of Fe-based Fischer-Tropsch catalyst: Deciphering the role of H2O[J]. Journal of Fuel Chemistry and Technology,
2020, 48(1): 63-74.
Received Date:
24 September 2019 Revised Date:
27 November 2019 Available Online:
01 January 2020
Abstract:
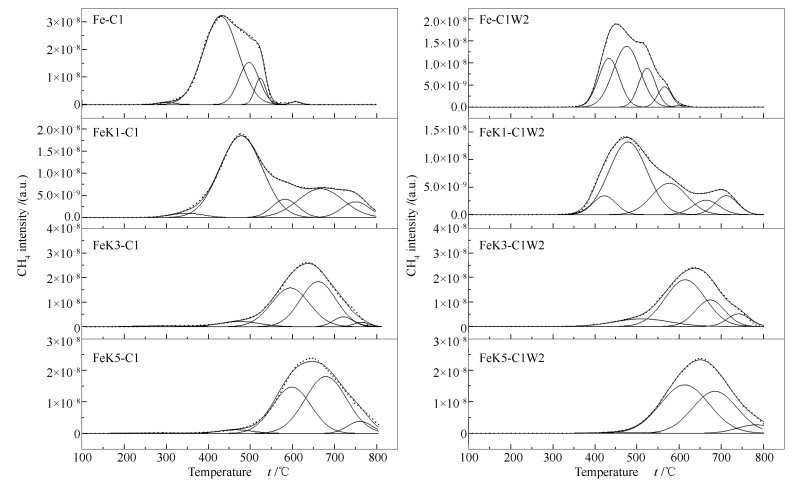
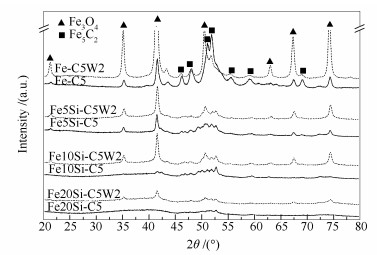
The effect of carburization and reduction degree on H2O oxidation behaviour for the iron carbides in Fe-based FTS catalyst were firstly investigated using a combination method including X-ray diffraction (XRD), Raman and temperature-programmed-hydrogenation (TPH). The relationship between carbon species transformation and H2O oxidation behaviour of iron carbides was investigated simultaneously. Based on these observations, the influence of typical promoters like K and SiO2 on the structure and H2O oxidation behaviour of Fe-based FTS catalysts was further studied. The results indicated that, for the iron catalyst, the stability of iron carbides against H2O oxidation was increased with the increase of iron carbides content, and the H2O oxidation process led to the formation of more graphitic carbon. The carburization ability was effectively enhanced when certain amount of K promoter was incorporated. Addition of K into Fe-based FTS catalyst increased the number of graphitic carbons, which increased the stability of iron carbides toward H2O oxidation ultimately. It was also found that promotion of SiO2 greatly degraded the carburization degree of the catalyst, while their tendency to be oxidized to form Fe3O4 in the H2O atmosphere was obviously hindered.
Peak area of carbon deposition via Raman /(×104 a.u.)
Increase ratio of carbon deposition after oxidation by H2Ob /%
Fe5C2
Fe3O4
Fe5C2
Fe3O4
Fe-C1
36.16
63.85
30.92
85.51
7.65
21.23
1.66
4.42
130.77
Fe-C1W2
5.24
94.75
2.75
25.83
0.87
10.18
Fe-C3
68.02
31.98
41.13
60.47
9.04
20.82
1.37
5.23
57.17
Fe-C3W2
26.89
73.11
4.26
26.97
0.96
8.22
Fe-C5
84.30
15.70
49.31
58.49
10.48
17.74
1.41
5.52
7.25
Fe-C5W2
34.99
65.02
7.40
23.31
1.08
5.92
a: calculated as: (FeCx content before oxidation -FeCx content after oxidation)/ FeCx content before oxidation ×100%; b: calculated from (area of deposited carbon after oxidation-area of deposited carbon before oxidation)/ area of deposited carbon before oxidation ×100%
温晓东, 杨勇, 相宏伟, 焦海军, 李永旺. 费托合成铁基催化剂的设计基础:从理论走向实践[J]. 中国科学:化学,
2017,47,(11): 1298-1311.
WEN Xiao-dong, YANG Yong, XIANG Hong-wei, JIAO Hai-jun, LI Yong-wang. The design principle of iron-based catalysts for fischer-tropsch synthesis:from theory to practice[J]. Sci Sin Chim,
2017, 47(11):
1298-1311.
[2]
ZHANG Q H, KANG J C, WANG Y. Development of novel catalysts for Fischer-Tropsch synthesis:Tuning the product selectivity[J]. ChemCatChem,
2010, 2(9):
1030-1058.
doi: 10.1002/cctc.201000071
[3]
张成华, 杨勇, 陶智超, 李廷真, 万海军, 相宏伟, 李永旺. Cu、K助剂对FeMn/SiO2催化费托合成的影响[J]. 物理化学学报,
2006,22,(11): 1310-1316.
ZHANG Cheng-hua, YANG Yong, TAO Zhi-chao, LI Ting-zhen, WAN Hai-jun, XIANG Hong-wei, LI Yong-wang. Effects of Cu and K on Co-precepitated FeMn/SiO2 catalysts for Fischer-Tropsch synthesis[J]. Acta Phys-Chim Sin,
2006, 22(11):
1310-1316.
[4]
YANG Y, XIANG H W, TIAN L, WANG H, ZHANG C H, TAO Z C, XU Y Y, ZHONG B, LI Y W. Structure and Fischer-Tropsch performance of iron-manganese catalyst incorporated with SiO2[J]. Appl Catal A:Gen,
2005, 284(1):
105-122.
[5]
ARGYLE M D, BARTHOLOMEW C H. Heterogeneous catalyst deactivation and regeneration:A review[J]. Catalysts,
2015, 5:
145-269.
doi: 10.3390/catal5010145
[6]
DRY M E, SHINGLES T, BOSHOFF L J, BOTHA C S H. Factors influencing the formation of carbon on iron Fischer-Tropsch catalysts:Ⅱ. The effect of temperature and of gases and vapors present during Fischer-Tropsch synthesis[J]. J Catal,
1970, 17(3):
347-354.
doi: 10.1016/0021-9517(70)90110-7
[7]
SARKAR A, SETH D, DOZIER A K, NEATHERY J K, HAMDEH H H, DAVIS B H. Fischer-Tropsch synthesis:Morphology, phase transformation and particle size growth of nano-scale particles[J]. Catal Lett,
2007, 117(1/2):
1-17.
[8]
MANSKER L D, JIN Y, BUKUR D B, DATYE A K. Characterization of slurry phase iron catalysts for Fischer-Tropsch synthesis[J]. Appl Catal A:Gen,
1999, 186(1/2):
277-296.
[9]
DRY M E, HOOGENDOORN J C. Technology of the Fischer-Tropsch process[J]. Catal Rev,
1981, 23(1/2):
265-278.
[10]
NING W, KOIZUMI N, CHANG H, MOCHIZUKU T, ITOH T, YAMADA M. Phase transformation of unpromoted and promoted Fe catalysts and the formation of carbonaceous compounds during Fischer-Tropsch synthesis reaction[J]. Appl Catal A:Gen,
2006, 312(9):
35-44.
[11]
PENDYALA V R R, JACOBS G, MOHANDAS J C, LUO M S, HAMDEH H H, JI Y Y, RIBEIRO M C, DAVIS B H. Fischer-Tropsch Synthesis:Effect of water over iron-based catalysts[J]. Catal Lett,
2010, 140(3/4):
98-105.
[12]
THÜNE P, MOODLEY P, SCHEIJEN F, FREDRIKSSON H, LANCEE R, KROPF J, MILLER J, NIEMANTSVERDRIET J W. The effect of water on the stability of iron oxide and iron carbide nanoparticles in hydrogen and syngas followed by in situ X-ray absorption spectroscopy[J]. J Phys Chem C,
2012, 116(13):
7367-7373.
doi: 10.1021/jp210754k
[13]
SATTERFIELD C N, HANLON R T, TUNG S E, ZOU Z M, PAPAEFTHYMIOU G C. Effect of water on the iron-catalyzed Fischer-Tropsch synthesis[J]. Ind Eng Chem Pro Res Dev,
1986, 25(3):
407-414.
doi: 10.1021/i300023a007
[14]
QING M, YANG Y, WU B S, XU J, ZHANG C H, GAO P, LI Y W. Modification of Fe-SiO2 interaction with zirconia for iron-based Fischer-Tropsch catalysts[J]. J Catal,
2011, 279(1):
111-122.
doi: 10.1016/j.jcat.2011.01.005
[15]
BUTT J B. Carbide phases on iron-based Fischer-Tropsch synthesis catalysts part Ⅰ:Characterization studies[J]. Catal Lett,
1990, 7(1/4):
61-81.
[16]
TUINSTRA F, KOENIG J L. Raman spectrum of graphite[J]. J Chem Phys,
1970, 53(3):
1126-1130.
doi: 10.1063/1.1674108
[17]
NEMANICH R J, SOLIN S A. First- and second-order Raman scattering from finite-size crystals of graphite[J]. Phys Rev B,
2015, 20(2):
392-401.
TAN P H, ZHANG S L, KWOK T Y, HUANG F M, SHI Z J, ZHOU X H, GU Z N. Comparative Raman study of carbon nanotubes prepared by D.C. arc discharge and catalytic methods[J]. J Raman Spectrosc,
1997, 28(5):
369-372.
doi: 10.1002/(SICI)1097-4555(199705)28:5<369::AID-JRS107>3.0.CO;2-X
[20]
SHULTZ J F, HALL W K, SELIGMAN B, ANDERSON R B. Studies of the Fischer-Tropsch synthesis. XIV. Hägg iron carbide as catalysts[J]. J Am Chem Soc,
1955, 77(1):
213-221.
doi: 10.1021/ja01606a079
[21]
XU J, BARTHOLOMEW C H. Temperature-programmed hydrogenation (TPH) and in situ Mössbauer spectroscopy studies of carbonaceous species on silica-supported iron Fischer-Tropsch catalysts[J]. J Phys Chem B,
2005, 109(6):
2392-2403.
doi: 10.1021/jp048808j
[22]
MILLER D G, MOSKOVITS M. A study of the effects of potassium addition to supported iron catalysts in the Fischer-Tropsch reaction[J]. J Phys Chem,
1988, 92(21):
6081-6085.
doi: 10.1021/j100332a047
[23]
BONZEL H P, KREBS H J. Enhanced rate of carbon deposition during Fischer-Tropsch synthesis on K promoted Fe[J]. Surf Sci,
1981, 109(2):
L527-L531.
doi: 10.1016/0039-6028(81)90486-6
[24]
FERDI S, SING K S W, WEITKAMP J. Handbook of Porous Solids(vol.3)[M]. Germany:Wiley-VCH, 2002:1543-1591.
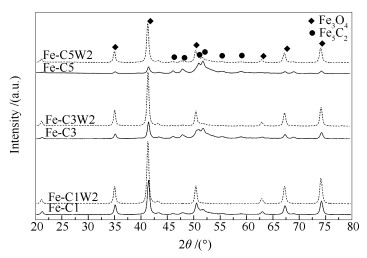
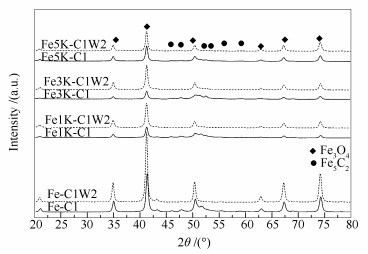
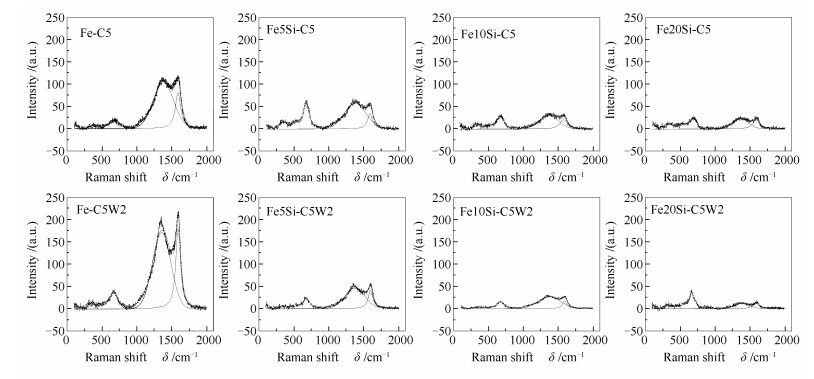
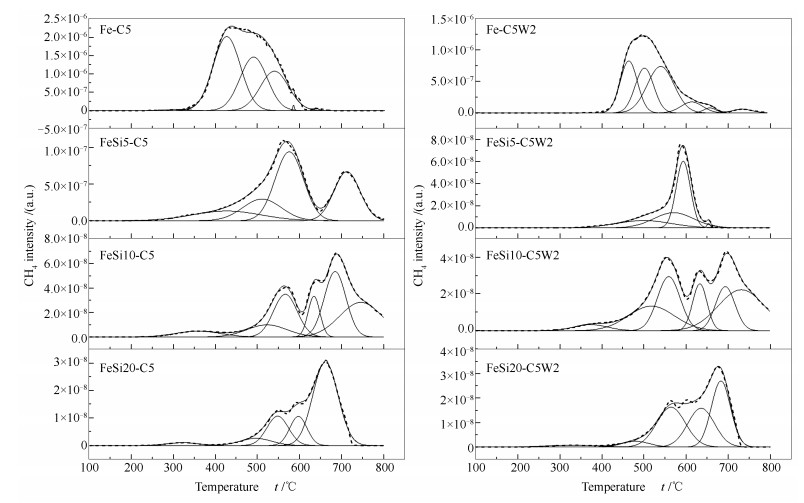
图 1
纯Fe催化剂不同时间碳化及水氧化后的XRD谱图
Figure 1
XRD patterns of Fe catalysts after carbonization (solid line) and oxidation (dotted line) for different times
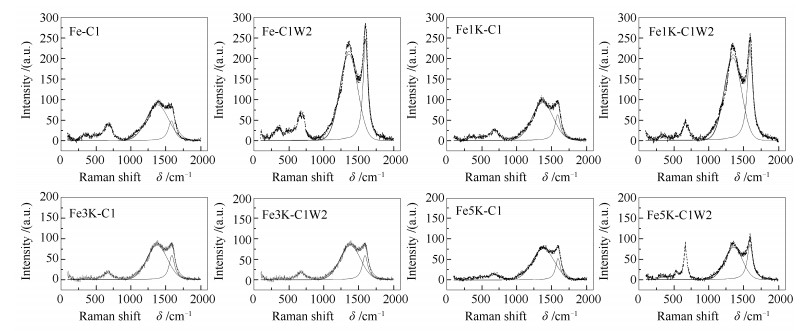
Table 1.
XRD and Raman results of Fe catalysts after carbonization and oxidization
Sample
Content wmol/%
FeCx phase shift wmol/%
Oxidation degreea D/%
Particle size d/nm
ID/IG
Peak area of carbon deposition via Raman /(×104 a.u.)
Increase ratio of carbon deposition after oxidation by H2Ob /%
Fe5C2
Fe3O4
Fe5C2
Fe3O4
Fe-C1
36.16
63.85
30.92
85.51
7.65
21.23
1.66
4.42
130.77
Fe-C1W2
5.24
94.75
2.75
25.83
0.87
10.18
Fe-C3
68.02
31.98
41.13
60.47
9.04
20.82
1.37
5.23
57.17
Fe-C3W2
26.89
73.11
4.26
26.97
0.96
8.22
Fe-C5
84.30
15.70
49.31
58.49
10.48
17.74
1.41
5.52
7.25
Fe-C5W2
34.99
65.02
7.40
23.31
1.08
5.92
a: calculated as: (FeCx content before oxidation -FeCx content after oxidation)/ FeCx content before oxidation ×100%; b: calculated from (area of deposited carbon after oxidation-area of deposited carbon before oxidation)/ area of deposited carbon before oxidation ×100%

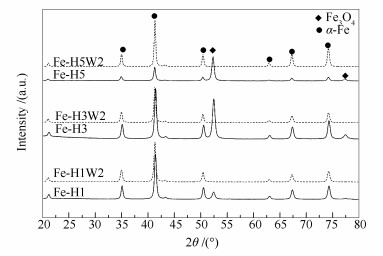
 下载:
下载:
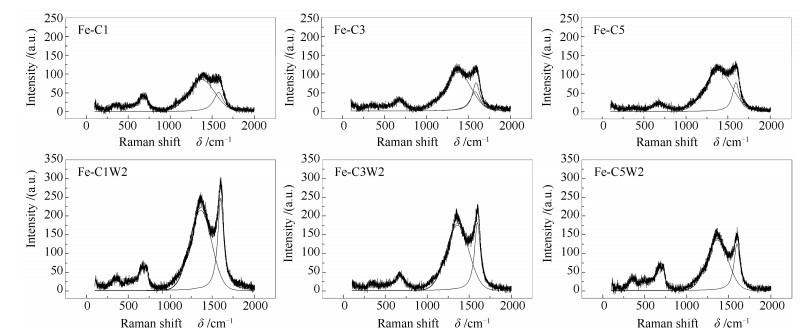
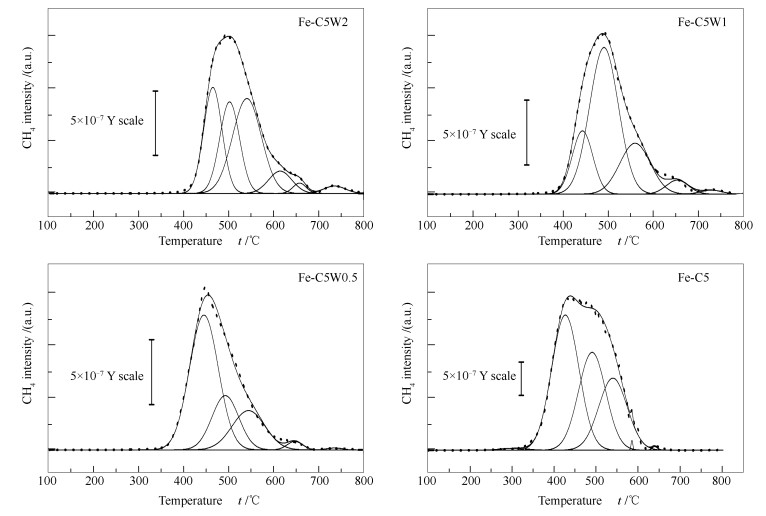

 下载:
下载:











