
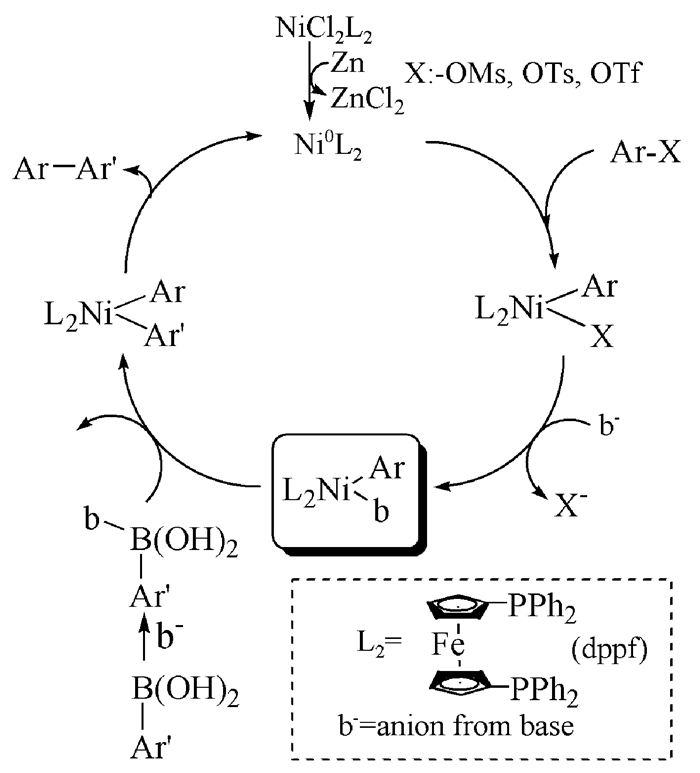
图式 1.
Ni催化的Suzuki偶联反应可能机理
Scheme 1.
Possible mechanism of Ni-catalyzed Suzuki coupling reaction

Suzuki偶联反应,又称Suzuki-Miyaura反应,是有机合成中构建C-C键的重要方法,最早用于合成Csp2-Csp2相连接的化合物,如联苯、多烯烃和苯乙烯的衍生物等。与传统均相催化反应相比,Suzuki偶联反应具有对水稳定、反应容忍度高(适用于多种活性官能团取代的底物)、副产物无毒且易于分离、高度的区域选择性等优点。近几年,随着研究的不断深入,在Suzuki偶联反应的产物类型(Csp2-Csp3、Csp3-Csp3相连接的化合物的合成)、反应机理、底物适用性、催化剂配体类型、添加剂和反应条件等方面均取得重大突破。
Suzuki偶联反应最经典的催化剂是Pd(II/0),例如Pd0(PPh3)4、PdⅡCl2、PdⅡCl2(dppf)、PdⅡ (OAc)2、PdⅡ (PPh3)2Cl2等,这些催化剂经常与具有较高催化活性的配体(如有机膦类、氮杂卡宾类等)配合使用,配体的共性是具有较强的供电子能力和较大的空间位阻,这是因为富电性的配体有利于氧化加成反应,而空间位阻大的配体有利于还原消除。
随着有机合成化学需求的不断增加,开发高效价廉的新催化剂和配体是研究Suzuki偶联反应的一个重要方向,非常规的Suzuki偶联反应(如Csp2-Csp3、Csp2-Csp3相连的碳碳键构建)引起科学家的广泛关注。相比于传统的Pd催化剂,一些廉价金属(Ni、Cu、Co等)价格便宜、稳定性高、选择性更好,且对惰性底物(如氯代芳烃和氟代芳烃)有显著活性,扩大了Suzuki偶联反应适用范围,是Suzuki偶联反应在催化剂选择和适用性方面的重要补充。已经有关于Ni催化Suzuki偶联反应的一些相关综述[1~6],本文主要介绍近十年Ni催化Suzuki偶联反应的发展历程和研究进展。为了使条理清晰,我们根据反应底物类型(键断裂的位置)为线索,并对Ni催化Suzuki偶联反应的机理进行归纳。
1995年,Percec等[7]发现,以1, 1′-二(二苯基膦)二茂铁(dppf)为配体的Ni催化剂(NiⅡCl2(dppf))在金属锌存在下可以温和地催化磺酸芳基酯类底物与苯硼酸反应,生成联苯化合物(式(1))。自此,Ni催化的Suzuki偶联反应逐步成为研究的热点。
|
|
(1) |
金属Ni与Pd处于同一主族,Ni催化剂之所以能在Suzuki偶联反应中崭露头角与其特殊性质有直接的关系。Ni的氧化还原态有Ni(0)/Ni(Ⅱ)和Ni(Ⅰ)/Ni(Ⅲ)两种,Ni比Pd的原子半径更小,因此,具有更强的亲核能力。从其可能的反应机理来看,Ni催化与Pd催化的Suzuki偶联反应机理相似,C-C键的构建都经历了氧化加成和还原消除两个核心步骤(图式 1)。
在有机合成中,C-C键的成键反应最难,因为C-C键的强度高,其键解离能(BDE)高达87.4kcal/mol,在没有催化剂存在时,反应需要较高的能量和更为苛刻的反应条件。过渡金属催化的C-C键的构建,生成的中间态M-C(Metal-Carbon)键化合物具有较低的能量,使反应更加容易进行(图 1)。研究表明,Ni-C键具有更低的键解离能,BDE为38.0~51.1kcal/mol,Pd-C键的BDE为48.3~55.2kcal/mol,这是Ni催化剂在构建C-C键反应中的主要优势。在催化循环过程中,Ni中间体的生成更为容易,其中还原消除所需的能量仅为16.8 kcal/mol,与氧化加成过程的能量差为4.1kcal/mol(氧化加成所需能量为20.9kcal/mol),即仅有4.1kcal/mol的能量是以热能的形式释放,而Pd催化剂在氧化加成和还原消除过程中却有19 kcal/mol能量的损耗(ΔE(RE)‡=24.9 kcal/mol,ΔE(OA)‡=43.9 kcal/mol)。同时在Ni催化过程中,M-C键的均裂所需要的能量也是最低的[8, 9]。因此,Ni催化剂在(构建C-C键)Suzuki偶联反应中具有更高的反应活性。
1995年,Percec等[7]报道了首例Ni催化的苯硼酸与甲基磺酸苯酯的Suzuki偶联反应。在THF溶液中,以10(mol)% NiⅡCl2(dppf)为催化剂、1.7倍量的Zn为还原剂、3.0倍量的K3PO4为碱,67℃加热的条件下,高选择性、中等产率得到联苯化合物,显示出Ni催化剂在Suzuki偶联反应中应用的可能性。
1996年,Saito等[10]报道了NiⅡCl2(dppf)/BuLi体系有效催化氯代芳烃参与Suzuki偶联反应。在80℃加热条件下,零价Ni作为催化剂可高产率地催化氯代芳烃和芳基硼酸发生偶联反应制得联苯化合物(式(2))。该反应官能团容忍度高,包含有吸电子或给电子基团的各类底物对反应影响不是很大,其中零价Ni通过NiⅡCl2(dppf)(10(mol)%)和n-BuLi“一锅法”反应来制备。值得说明的是,在Ni催化下,含有供电子基团的底物对电负性更加不敏感,相同底物被Ni催化剂催化的收率远高于Pd催化剂催化的收率。
|
|
(2) |
在反应中,Ni(Ⅱ)催化剂常用的配体一般为膦,Ni(Ⅱ)必须被还原成Ni(0),通常需要加入其他还原剂,如Zn、NaH、n-BuLi和DIBAH等。1997年,Indolese[11]发现,NiⅡCl2(dppf)/K3PO4催化体系不需要任何额外的还原剂(式(3)),在苯甲醚或者1, 4-二氧六环中即可高效顺利地进行偶联反应。
|
|
(3) |
2000年,Lipshutz等[12, 13]发现,非均相负载Ni0/C(用NiⅡ(NO3)2和碳的混合物用4倍量的n-BuLi还原制备)可以催化芳基卤代物进行Suzuki偶联反应(式(4))。该反应条件相对温和,底物中包含的醛基、氰基、酮等均不受影响,因此,这个反应体系当时被认为是同类反应中最为“绿色”的。
|
|
(4) |
2004年,Tang等[14]发现Ni0(COD)2/PCy3体系可以在室温下催化芳香磺酸酯和苯硼酸的偶联反应(式(5))。由于Ni0(COD)2的活性太高,反应需要在手套箱中进行,为了稳定催化剂中的Ni(0),反应中经常需要加入膦配体[15, 16]、红菲绕啉[17]和咪唑盐[18]。该体系对惰性的氯代芳烃的Suzuki偶联反应具有较好的活性,同时,对包含各类取代基的惰性氯代芳烃都有较好的适用性。
|
$ \begin{gathered} {\text{Ar}} - {\text{OS}}{{\text{O}}_2}{\text{A}}{{\text{r}}^{\prime \prime }} + {\text{A}}{{\text{r}}^\prime } - {\text{B}}{({\text{OH}})_2} \hfill \\ \xrightarrow[{{\text{THF }}, {\text{r}}.{\text{t}}.{\text{8}}\sim 12{\text{h}}}]{{\begin{array}{*{20}{c}} {{{\text{K}}_3}{\text{P}}{{\text{O}}_4}} \\ {{\text{Ni}}{{({\text{COD}})}_2}(3({\text{mol}})\% ){\text{PC}}{{\text{y}}_3}(12({\text{mol}})\% )} \end{array}}}{\text{Ar}} - {\text{A}}{{\text{r}}^\prime } \hfill \\ \;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;86\% \sim 99\% \hfill \\ \end{gathered} $ |
(5) |
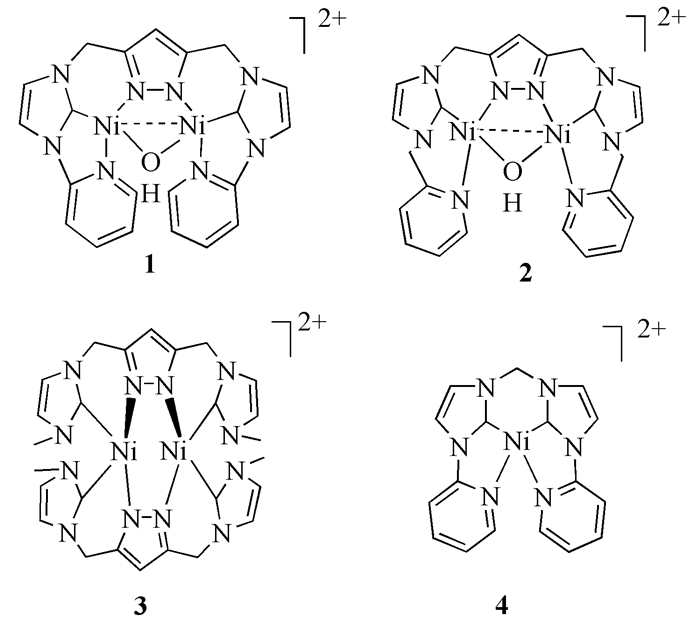
随着研究的不断深入,Ni催化Suzuki偶联反应的催化剂种类也得到不断增加,NiⅡCl2(dppf)、NiⅡBr2(dppf)、NiⅡCl2(dppb)、NiⅡCl2(PPh3)2和Ni0(COD)2等都是非常具有潜力的催化剂。2008年,N杂环配体Ni催化剂(图式 2)一经发现,就因其稳定性和易于操作性,得到众多学者的关注。Zhou等[19]发现催化剂1和2在催化对苯基氯代物和苯硼酸的偶联反应中产率较好(78%~98%)。随后大量的氮杂卡宾类配体的Ni催化剂被逐渐开发,应用也日益广泛,其中,催化剂4对除氯代物和溴代物以外的芳基磺酸酯等也具有很好的催化效果[20, 21]。
芳基卤代物是Suzuki偶联反应的重要底物,其中芳基氯代物和芳基氟代物发生氧化加成反应的活性很低,很少作为Suzuki偶联反应的备用底物。研究表明,当氯代芳烃的两个邻位都有取代基时,由于C-Cl键的稳定及空间位阻过大,很难氧化加成,导致反应无法进行;氟苯中C-F键的键能达到154kcal/mol,故氟代芳烃和烯基氟代物也很少作为C-C构建反应的备用底物[22]。随着研究的深入,对惰性卤代烃性质的认识不断提升,例如,大位阻和供电子能力强的配体能有效促进氯代芳烃进行Suzuki偶联反应。与此同时,惰性卤代芳烃与芳基硼化物的Suzuki偶联反应的催化剂选用范围也得到扩大,Au[23]、Ru[24]、Ni催化剂等相继得到开发,这些催化剂能有效促进反应的进行,尤其是Ni催化剂表现出对氯代芳烃类底物特有的活性,也为惰性卤代烃的Suzuki偶联反应提供了新的廉价选择。
氟代芳烃在偶联反应中的活性很低。Schaub等[25]发现,全氟取代苯与芳基硼酸在Ni催化剂的催化下可以发生Suzuki偶联反应(式(6))。这类反应常需加入添加剂N-杂环卡宾(NHC)用来稳定配体{[Ni20(i-Pr2Im)4(COD)] (i-Pr2Im:1, 3-双(异丙基)咪唑-2-亚基)},反应位点一般在三氟甲基和氟苯基取代基的对位。
|
|
(6) |
2011年,Tobisu等[26]在氟苯化合物和苯基硼酸酯的Suzuki偶联反应中,首次加入金属氟化物ZrF4和TiF4(式(7))。这种双金属催化体系打破了此类反应只能选择含有超强吸电子的氟苯底物(比如邻硝基氟苯和全氟苯)的局限性,成功将底物进行了扩展,使含有一般吸电子基团的底物(酮、酯和三氟甲基取代的氟苯、氟代萘、氟代喹啉等)在Ni催化剂的催化下也可以顺利进行Suzuki偶联反应。定位官能团取代的氟苯与金属作用生成环金属化的中间体能有效促进氧化加成反应,使反应活性显著提高。该双金属催化剂体系同样适用于定位基为sp2杂化氮原子的底物,如吡啶、吡唑和噁唑等。2015年,Xiong等[27]对2, 2-二氟烯基苯衍生物底物的Suzuki反应的研究取得进展,高选择性地得到Z-氟取代的苯乙烯化合物。
|
|
(7) |
同时,Ni催化Suzuki偶联反应的绿色方法也得到不断开发。Ramgren等[28]报道了使用绿色溶剂(2-甲基四氢呋喃和叔丁醇)的Suzuki偶联反应,采用市售NiⅡCl2(PCy3)2作为催化剂,高产率地得到联苯类化合物(式(8)),该反应催化剂用量低、底物普适性好,杂环类底物同样适用,具有较好的应用前景;Fan等[29]用NiⅡ-(α-aryl)配合物[Ni0(PPh3)2(1-naphthyl)Cl]作为催化剂前体,在未用有机金属试剂和还原剂处理的情况下,成功合成了联苯类化合物;Handa等[30]发现,原位生成的Ni纳米粒子可以作为Suzuki偶联反应的催化剂,该催化剂活性高并且性质稳定,反应可以在纯水中平稳进行。这些方法的开发使得Ni催化的Suzuki偶联反应极为简化,扩大了应用范围。
|
|
(8) |
2008年,Fu等[31]发展了Ni0(COD)2/6类催化体系,成功实现了非活性的烷基化溴化物与烷基化硼化物的不对称反应,ee值最高达到90%(式(9))。该反应揭示了在Ni催化剂作用下,惰性卤代物亲电试剂底物具备进行不对称构建碳碳键的能力。
|
|
(9) |
受到Ni0(COD)2/6催化体系的启发,将苯基替换为氨基酯类、磺胺基、砜类[32]、胺类[33]底物的不对称Suzuki偶联反应相继得到开发,底物中杂原子的增加使得催化剂的控制性更好,ee值得到小幅提高。进一步研究该体系的影响因素发现,底物结构对结果有重要影响。以苯胺类取代的底物为例,当增加N原子与卤原子取代碳原子之间的碳链长度后,ee值大为降低,底物增加一个碳原子,ee值从89%降到5%。
|
|
(10) |
2013年,Fu等[34]发展了不活泼的三取代碳卤代物的Suzuki偶联反应(式(11))。采用常见的NiⅡBr2·DME(二乙二醇二甲醚)/4, 4′二叔丁基-2, 2′-联吡啶体系(均为商品化试剂)为催化剂,成功构建了全新的Csp3-Csp2键,展现了不活泼的三取代碳卤代物在Suzuki偶联反应中的应用前景,该方法是四级碳类化合物的合成策略的补充,并且产物的构型得到保留。目前,对该反应的机理认识还不是很透彻,初步认为该反应在氧化加成时经历了自由基历程。
|
|
(11) |
2017年,Huang等[35]发展了一种Ni催化不对称Suzuki偶联反应(式(12))。为了保证反应的立体选择性,他们巧妙地选用苯硼酸频哪醇酯与正丁基锂生成高活性的亲核试剂,使得反应能在较低温度的条件下进行,高效构建出包含三氟甲氧基的手性中心。反应的底物官能团性容忍性较好,间位和对位含有各种电性取代基的底物均可以顺利地发生反应,反应的产率和对映选择性都较高。
|
|
(12) |
磺酸类衍生物是Suzuki偶联反应中最重要的亲电底物。当HBF4存在时,以t-BuOH/H2O(1:1)为溶剂、2-萘酚类底物可以在10(mol)% Ni0(COD)2/20 (mol)% PCy3催化体系下进行Suzuki偶联反应,不同类型的底物活性顺序为OMs>OTs≈OSO2NMe2>OPiv>OBoc>OCONE2>OBz≫OMe=0[36],其中磺酸类衍生物底物在Suzuki偶联反应中的活性较高,在研究Suzuki偶联反应机理时具有一定的优势。
|
|
(13) |
2011年,Leowanawat等[37]报道了Ni0(COD)2/PCy3催化体系能够催化苯基甲磺酸酯与苯基新戊二醇硼酸酯衍生物的Suzuki偶联反应(式(13))。该催化剂体系在Suzuki偶联反应中应用广泛,各种苯环上的取代基均不影响反应结果。Quasdorf等[38]发现,在110℃下,Ni(Ⅱ)催化剂可以催化N,N-二甲基氨基磺酸芳基酯类化合物发生Suzuki偶联反应。研究表明,Ni催化剂对C-O衍生物的亲电试剂具有更高活性[39]。一般情况下,Suzuki偶联反应中的Ni催化剂在反应体系中有三种氧化形态,即Ni(0)、Ni(Ⅰ)或者Ni(Ⅱ)[40, 41]。Guard等[42]通过研究与dppf相互作用的三种形态Ni催化剂发现,无论哪种形式的Ni催化剂加入,反应体系中都会有Ni(Ⅰ)生成。2017年,他们发现反应中苯基氨基磺酸酯的金属转移过程与Ni的歧化反应存在竞争关系[43],如式(14)所示,在催化剂的歧化反应中明确观察到Ni(Ⅰ)-胺磺酰基和Ni(Ⅰ)-芳基配合物存在。他们认为无论是从Ni(Ⅰ)的来源还是其在催化循环中的作用来看,均对该类反应的进行是不利的。Ni(Ⅰ)在反应中的作用是进一步深入研究Ni催化Suzuki偶联反应的关键之一。2018年,Payard等[44]对硼到镍的金属转移过程进行了详细研究,发现在反应体系中碱和硼酸的比例是调控双核(Ni)配体生成的重要指标,该双核(Ni)配体阻碍了硼到镍的金属转移过程,使催化剂的活性大为降低。
|
|
(14) |
2008年,Tobisu等[45]发现在Ni0(COD)2/PCy3体系催化下,苯基甲基醚可以与苯硼酸酯发生Suzuki偶联反应,得到联苯化合物(式(15))。该反应对各类芳基甲基醚和苯硼酸化合物均有很好的容忍度。Guo等[46, 47]随后对硼酸偶联底物进行深度扩展,开发出烷基硼酸插入C-OMe键的反应,克服了底物的β氢消除,成功构建出Csp2-Csp3键底物。
|
|
(15) |
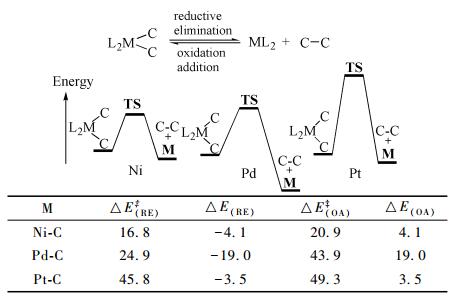
2009年,Yu等[48]发现苯基乙基醚类底物同样有较好的反应效果。2014年,Tobisu等[49]开发了Ni0/ICy催化剂体系,该催化剂通过Ni0(COD)2与ICy结合制得,可催化苄基甲基醚与苯硼酸类化合物发生偶联反应得到亚甲基桥连的双苯基化合物。随后的研究表明,这类催化剂在C-O键断链反应中具有较强的活性,在双联硼酸酯存在下苯甲醚直接发生偶联反应得到联苯化合物[50](图式 3)。
2017年,Wu等[51]发展了一类新型膦配体9,该配体与Ni催化剂相互作用使其活性显著提升,能高效催化苄基的缩醛底物与芳基硼氧烷的Suzuki偶联反应(式(16)),通过测量和模拟分析,发现此类配体尺寸与Ni匹配度较高,Ni原子半径更小,这种催化剂配体体积小、锥角大,更有利于提高Ni催化剂的催化活性。该配体的开发为之后的Ni催化剂配体的设计提供了参考模板,作者提出的远程位阻概念为Ni催化剂配体的设计提供了新的方向,具有重大指导意义。
2007年,Hansen等[52]在Nan等[53]工作的基础上发展了一种多功能的Suzuki偶联反应(式(17)),反应底物为非活性的烯基磷酸酯和苯硼酸,得到苯基取代的烯基类化合物。该反应有较好的容忍度,很多类型的烯基磷酸酯的衍生物和苯基硼酸衍生物可以进行反应。
|
|
(16) |
|
|
(17) |
2010年,Zhao等[54]发现芳基磷酰胺化合物可以作为Suzuki偶联反应的亲电试剂(式(18))。与各类苯硼酸衍生物发生反应,高产率得到联苯化合物。这种苯基“bop”亲电试剂由苯酚和磷酰氯制得,为杂环底物的Suzuki偶联反应提供很好的选择,3位被羟基取代或者4位被羟基取代的吡啶在该反应中同样高效。
|
|
(18) |
2011年,Chen等[55]发现,NiⅡ(PCy3)2/Cl2可以很好地将惰性的磷酸苯基酯活化,使其与各种苯硼酸反应得到联苯化合物(式(19))。Pd(0)和Pd(Ⅱ)催化剂并不能催化此类反应,说明Ni催化剂在活化C-O键断裂时具有比Pd催化剂更好的效果。
|
|
(19) |
苯基酯充当过渡金属催化的Suzuki偶联反应的亲电底物相比卤代物具有更加环境友好的特点,但是由于酯基较好的稳定性和过高的键能,偶联反应中氧化加成难度很大。
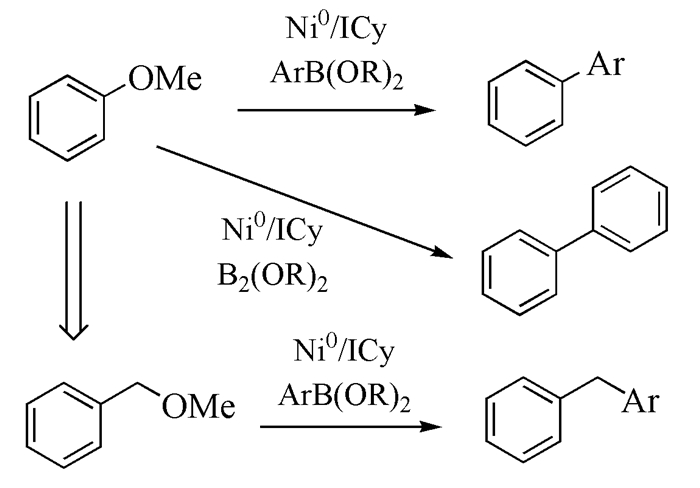
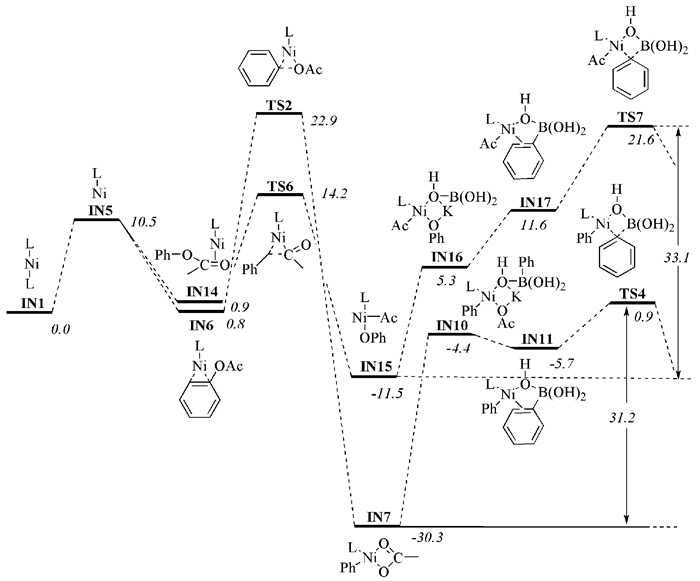
2008年,Quasdorf等[56]和Guan等[57]先后报道了苯酚酯与苯硼酸类化合物参与的Suzuki偶联反应(图式 4)。这两个反应策略除了苯硼酸类化合物不同,其余条件基本相似,三甲基乙酸苯酯均被认为是最佳苯基酯反应底物、反应催化剂为NiⅡCl2/(PCy3)2、碱都选择K3PO4。Li等[58]和Hong等[59]对该反应的机理进行研究发现,该反应同样是由三个阶段构成,即氧化加成、过渡金属转移和还原消除。Ar-OAc键的氧化加成在Ni(0)单膦配体催化剂中进行(以往在Ni(0)双膦配体催化剂中进行),需要能量为+22.9kcal/mol,过渡金属转移需要能量+31.2kcal/mol,而ArO-Ac键进行氧化加成需要+14.2 kcal/mol的能量,过渡金属转移需要能量+33.1 kcal/mol(图 2)。相比之下,ArO-Ac键发生氧化加成需要的能量较小,过程是可逆过程,过渡金属转移不容易进行(需要能量高)。因此,反应优先在Ar-OAc键处发生,与实验结果相符。
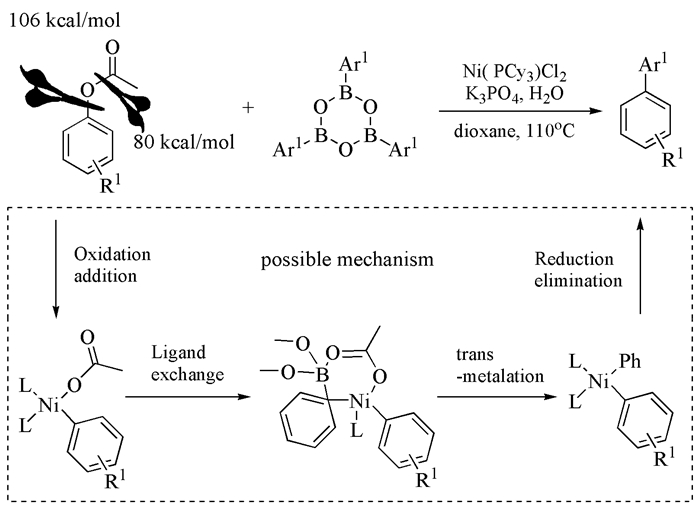
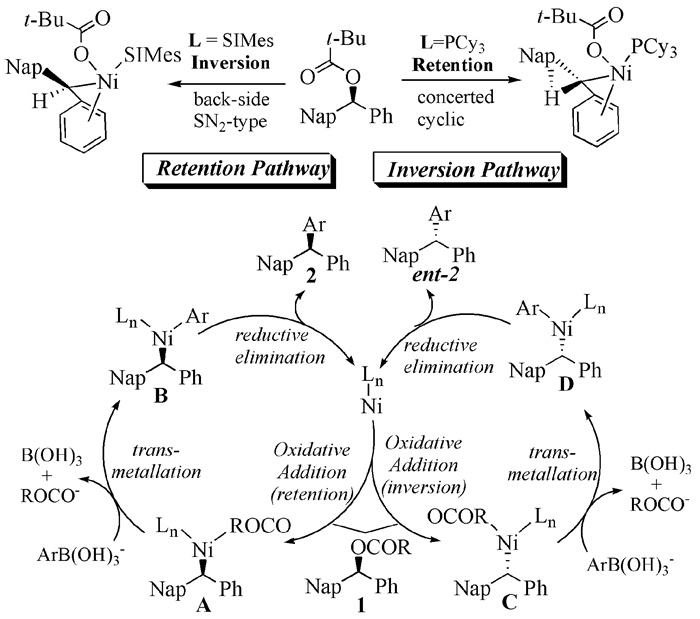
2013年,Basch等[60]发现,Ni0(COD)2/甲醇钠催化体系可以在Suzuki偶联反应中实现手性苄基三甲基乙酸酯构型翻转,ee值达到98%以上(式(20))。该反应不仅为二苯基取代和三苯基取代化合物的不对称合成提供了新的路径,而且让人们认识到Ni催化剂在不对称Suzuki偶联反应中的应用潜力。与此同时,Zhou等[61]对底物进行扩展,发现底物构型变化与配体结构有直接作用,三环己基膦配体使反应的构型得到保留,而NHC类配体则得到构型反转的产物(式(21))。
|
|
(20) |
|
|
(21) |
Harris等[62]对该反应的机理进行了研究,通过密度泛函理论(DFT)计算研究配体对反应构型的影响,发现催化剂与亲电底物的氧化加成对反应有决定性作用,加成过程中遵循SN2机理类型,反应的构型也在此处引入,不同的配体导致不同类型底物-Ni-配体的扭转角不同,影响硼化物进入的方向,并通过计算重现了两种配体在C-O键断裂中的立体专一性。对PCy3配体而言,手性保持的环状协同过渡态比SN2型的断裂方式具有1.1kcal/mol的优势;而对于SIMes配体而言,手性反转的SN2型断裂方式却比环状的协同过渡态具有1.6kcal/mol的优势(图式 5)。
2016年,Shi等[63]报道了首例金属Ni催化的芳香酰胺作为亲核试剂合成联苯的Suzuki偶联反应(式(22))。他们推测,该反应首先是Ni催化剂活化酰胺类底物中的N-C键,再经过脱羰基,最后得到联苯类化合物。该反应普适性好,底物中含有各类官能团均能得到很好的反应结果。
|
|
(22) |
Wang等[64]发现一级和二级脂肪羧酸被TCNHPI(N-羟基-四氯邻苯二甲酰亚胺)活化后具备了较强的亲核能力,实验结果也表明该策略可作为合成芳基脂肪羧酸取代的产物的可靠方法(式(23)),初步认为该反应经历了自由基历程。
|
|
(23) |
2018年,Rueping等[65, 66]开发了不同配体的Ni催化剂,高选择性地对C(acyl)-O键和C(aryl)-C键进行切断反应(式(24)),通过简单的切换配体,酯的底物可以有效地转化为烷基化芳烃和芳基酮,对各类取代基的底物均具有较好适用性。研究表明,Ni催化剂含有双齿配体时更容易使C(aryl)-C键发生氧化加成,而单配位的膦配体容易使C(acyl)-O键发生氧化加成。
|
|
(24) |
Garg等[67, 68]报道了Ni催化的脂肪酰胺衍生物作为亲核试剂的Suzuki偶联反应(式(25)),打破了酰胺类底物活性过低,从C-N键断裂来构建新C-C键非常困难的认知。研究结果显示,配体12、13、14、15与Ni形成的催化剂可以有效催化该类酰胺衍生物发生偶联反应,底物容忍度很高,对合成脂肪酮类化合物提供了新的选择;而且芳香性的苯基酰胺衍生物同样可以在Ni/SIPr催化剂体系下高产(96%)地得到苯基酮类化合物。Xu等[69]对该反应的机理进行了深入研究,发现体系中的水有提供质子氢的作用,质子转移也是该反应的决速步。
|
|
(25) |
Zhang等[70]发展了一种以烷基胺为底物的Suzuki偶联反应(式(26))。该反应过程中不活泼的烷基胺转化为活性的吡啶盐,实现C-N键活化,使其具备能与催化剂匹配的亲电能力,该盐在催化剂NiⅡ(OAc)2的作用下,与苯硼酸发生Suzuki偶联反应。此反应容忍度较好,一级和二级脂肪胺均可以发生反应,其反应机理可能是Ni(Ⅰ)/Ni(Ⅲ)催化循环。
|
|
(26) |
2018年,Ariki等[71]在Fu等[34]和Zhou等[72]工作的基础上,将底物类型进行了扩展,发现用苯基砜或者烯丙基砜与苯硼酸类化合物在Ni催化剂作用下可以发生Suzuki偶联反应(式(27))。该方法采用Wu等[51]开发的Ni催化剂配体,获得合成四级全碳化合物。应用该方法使得维生素D的合成路线得到大大的简化。
|
|
(27) |
本文以Ni催化的Suzuki偶联反应中亲电试剂底物的类型为线索,总结了各种类型Suzuki偶联反应(碳卤键断裂类、碳氧键断裂类、碳碳键断裂类、碳氮键断裂类和碳硫键断裂类)近十年的进展。Ni催化Suzuki偶联反应的研究发展主要集中在三个方面:(1)亲电试剂底物不断丰富,非常规Suzuki偶联反应的亲电试剂底物如氟代芳烃、Csp3-X底物、酰胺类底物等相继被开发出来;(2)反应的应用范围不断扩大,逐步成为构建含有Csp2-Csp3、Csp3-Csp3键化合物的有效方法;(3)机理研究取得突破,发现各类氧化还原态的Ni在Suzuki偶联反应中的重要作用,为日后机理探索、反应设计(配体和催化剂设计)奠定了坚实的理论基础。
综上所述,Ni催化的Suzuki偶联反应仍然处于方法发展阶段,未来Ni催化Suzuki偶联反应的研究将会集中在机理探究、新型催化剂和配体设计、多类型碳碳键构建(Csp2-Csp3、Csp3-Csp3)以及广泛底物的不对称Ni催化Suzuki偶联反应等方面。
B M Rosen, K W Quasdorf, D A Wilson et al. Chem. Rev., 2011, 111:1346~1416. doi: 10.1021/cr100259t
R Jana, T P Pathak, M S Sigman. Chem. Rev., 2011, 111:1417~1492. doi: 10.1021/cr100327p
F S Han. Chem. Soc. Rev., 2013, 42:5270~5298. doi: 10.1039/c3cs35521g
M Irene, N Oscar. Molecules, 2015, 20:7528 doi: 10.3390/molecules20057528
T Ryosuke, M Kei, Y Junichiro. Chem. Soc. Rev., 2017, 46:5864~5888. doi: 10.1039/C7CS00182G
陈国军, 杜建时.有机化学, 2014, 34(01):65~80. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=yjhx201401005
V Percec, J Y Bae, D H Hill. J. Org. Chem., 1995, 60:1060~1065. doi: 10.1021/jo00109a044
S A Macgregor, G W Neave, C Smith. Faraday Discuss, 2003, 124:111~127. doi: 10.1039/B212309F
V P Ananikov, D G Musaev, K Morokuma. Organometallics, 2005, 24:715~723. doi: 10.1021/om0490841
S Saito, M Sakai, N Miyaura. Tetrahed. Lett., 1996, 37:2993~2996. doi: 10.1016/0040-4039(96)00482-0
A F Indolese. Tetrahed. Lett., 1997, 38:3513~3516. doi: 10.1016/S0040-4039(97)00707-7
B H Lipshutz, J A Sclafani, P A Blomgren. Tetrahedron, 2000:2139~2144. https://www.sciencedirect.com/science/article/pii/S0040402099010960
B H Lipshutz, W Chrisman, S Tasler et al. J. Org. Chem., 2003, 68:1177~1189. doi: 10.1021/jo020296m
Z Y Tang, Q S Hu. J. Am. Chem. Soc., 2004, 126:3058~3059. doi: 10.1021/ja038752r
Z Y Tang, S Spinella, Q S Hu. Tetrahed. Lett., 2006, 47(14):2427~2430. doi: 10.1016/j.tetlet.2006.01.145
D A Wilson, C Wilson J, B M Rosen et al. Org. Lett., 2008, 10:4879~4882. doi: 10.1021/ol801972f
J Zhou, G C Fu. J. Am. Chem. Soc., 2004, 126:1340~1341. doi: 10.1021/ja039889k
J Liu, M J Robins. Org. Lett., 2005, 7:1149~1151. doi: 10.1021/ol050063s
Y B Zhou, Z X Xi, W Z Chen et al. Organometallics, 2008, 27:5911~5920. doi: 10.1021/om800711g
J I Kuroda, K Inamoto, K Hiroya et al. Eur. J. Org. Chem., 2009, (14):2251~2261. doi: 10.1002/ejoc.200900067
K Inamoto, J Kuroda, E Kwon et al. J. Organomet. Chem., 2009, 694:389~396. doi: 10.1016/j.jorganchem.2008.11.003
H Amii, K Uneyama. Chem. Rev., 2009, 109:2119~2183. doi: 10.1021/cr800388c
J Han, Y Liu, R Guo. J. Am. Chem. Soc., 2009, 131:2060~2061. doi: 10.1021/ja808935n
T Hoshi, T Honma, A Mori et al. J. Org. Chem., 2013, 78:11513~11524. doi: 10.1021/jo402089r
T Schaub, M Backes, U Radius. J. Am. Chem. Soc., 2006, 128:15964~15965. doi: 10.1021/ja064068b
M Tobisu, T Xu, T Shimasaki et al. J. Am. Chem. Soc., 2011, 133:19505~19511. doi: 10.1021/ja207759e
Y Xiong, T Huang, X F Ji et al. Org. Biomol. Chem., 2015, 13:7389~7392. doi: 10.1039/C5OB01016K
S D Ramgren, J Hie, Y X Ye et al. Org. Lett., 2013, 15(15):3950~3953. doi: 10.1021/ol401727y
X H Fan, L M Yang. Eur. J. Org. Chem., 2011, (8):1467~1471.
S Handa, E D Slack, B H Lipshutz. Angew. Chem. Int. Ed., 2015, 54:11994~11998. doi: 10.1002/anie.201505136
B Saito, G C Fu. J. Am. Chem. Soc., 2008, 130:6694~6695. doi: 10.1021/ja8013677
A Wilsily, F Tramutola, N A Owston et al. J. Am. Chem. Soc., 2012, 134:5794~5797. doi: 10.1021/ja301612y
Z Lu, A Wilsily, G C Fu. J. Am. Chem. Soc., 2011, 133:8154~8157. doi: 10.1021/ja203560q
S L Zultanski, G C Fu. J. Am. Chem. Soc., 2013, 135:624~627. doi: 10.1021/ja311669p
W C Huang, X L Wan, Q L Shen. Angew. Chem., 2017, 129(39):12148~12151. doi: 10.1002/ange.201706868
G A Molander, F Beaumard. Org. Lett., 2010, 12:4022~4025. doi: 10.1021/ol101592r
P Leowanawat, N Zhang, A M Remerita et al. J. Org. Chem., 2011, 76:9946~9955. doi: 10.1021/jo202037x
K W Quasdorf, A F Aurora, P Liu et al. J. Am. Chem. Soc., 2011, 133:6352~6363. doi: 10.1021/ja200398c
B M Rosen, K W Quasdorf, D A Wilson et al. Chem. Rev., 2011, 111:1346~1416. doi: 10.1021/cr100259t
N D Schley, G C Fu. J. Am. Chem. Soc., 2014, 136:16588~16593. doi: 10.1021/ja508718m
J Cornella, G B Enrique, R Martin. J. Am. Chem. Soc., 2013, 135:1997~2009. doi: 10.1021/ja311940s
L M Guard, B M Mohadjer, G W Brudvig et al. Angew. Chem. Int. Ed., 2015, 54:13352~13556. doi: 10.1002/anie.201505699
B M Mohadjer, A Nova, D Balcells et al. J. Am. Chem. Soc., 2017, 139:922~936. doi: 10.1021/jacs.6b11412
P A Payard, L A Perego, I Ciofini et al. ACS Catal., 2018, 8:4812~4823. doi: 10.1021/acscatal.8b00933
M Tobisu, T Shimasaki, N Chatani. Angew. Chem. Int. Ed., 2008, 47:4866~4869. doi: 10.1002/(ISSN)1521-3773
L Guo, X Q Liu, C Baumann et al. Angew. Chem. Int. Ed., 2016, 55:15415~15419. doi: 10.1002/anie.v55.49
L Guo, C C Hsiao, H F Yue et al. ACS Catal., 2016, 6(7):4438~4442. doi: 10.1021/acscatal.6b00801
D G Yu, M Yu, B T Guan et al. Org. Lett., 2009, 11:3374~3377. doi: 10.1021/ol901217m
M Tobisu, A Yasutome, H Kinuta et al. Org. Lett., 2014, 16:5572~5575. doi: 10.1021/ol502583h
K Nakamura, M Tobisu, N Chatani. Org. Lett., 2015, 17:6142~6145. doi: 10.1021/acs.orglett.5b03151
K Wu, A G Doyle. Nat. Chem., 2017, 9:779~784. doi: 10.1038/nchem.2741
A L Hansen, J P Ebran, T M Gøgsig et al. J. Org. Chem., 2007, 72:6464~6472. doi: 10.1021/jo070912k
Y Nan, Z Yang. Tetrahed. Lett., 1999, 40:3321~3324. doi: 10.1016/S0040-4039(99)00517-1
Y L Zhao, Y Li, Y Li et al. Chem. Eur. J., 2010, 16:4991~4994. doi: 10.1002/chem.v16:17
H Chen, Z Huang, X Hu et al. J. Org. Chem., 2011, 76:2338~2344. doi: 10.1021/jo2000034
K W Quasdorf, X Tian, N K Garg. J. Am. Chem. Soc., 2008, 130:14422~14423. doi: 10.1021/ja806244b
B T Guan, Y Wang, B J Li et al. J. Am. Chem. Soc., 2008, 130:14468~14470. doi: 10.1021/ja8056503
Z Li, S L Zhang, Y Fu et al. J. Am. Chem. Soc., 2009, 131:8815~8823. doi: 10.1021/ja810157e
X Hong, Y Liang, K N Houk. J. Am. Chem. Soc., 2014, 136:2017~2025. doi: 10.1021/ja4118413
C H Basch, J Liao, J Xu et al. J. Am. Chem. Soc., 2017, 139:5313~5316. doi: 10.1021/jacs.7b02389
Q Zhou, H D Srinivas, S Dasgupta et al. J. Am. Chem. Soc., 2013, 135:3307~3310. doi: 10.1021/ja312087x
M R Harris, L E Hanna, M A Greene et al. J. Am. Chem. Soc., 2013, 135:3303~3306. doi: 10.1021/ja311783k
S C Shi, G R Meng, M Szostak. Angew. Chem. Int. Ed., 2016, 55:6959~6963. doi: 10.1002/anie.201601914
J Wang, T Qin, T G Chen et al. Angew. Chem. Int. Ed., 2016, 55:9676. doi: 10.1002/anie.201605463
A Chatupheeraphat, H H Liao, W Srimontree et al. J. Am. Chem. Soc., 2018, 140:3724~3735. doi: 10.1021/jacs.7b12865
L Guo, M Rueping. Acc. Chem. Res., 2018, 51:1185~1195. doi: 10.1021/acs.accounts.8b00023
T B Boit, N A Weires, J Kim et al. ACS Catal., 2018, 8:1003~1008. doi: 10.1021/acscatal.7b03688
N A Weires, E L Baker, N K Garg. Nat. Chem., 2016, 8:75~79. doi: 10.1038/nchem.2388
Z Y Xu, H Z Yu, Y Fu. Chem. Asian J., 2017, 12:1765~1772. doi: 10.1002/asia.v12.14
S Q Zhang, B L H Taylor, C L Ji et al. J. Am. Chem. Soc., 2017, 139:12994~13005. doi: 10.1021/jacs.7b04973
Z T Ariki, Y Maekawa, M Nambo et al. J. Am. Chem. Soc., 2018, 140:78~81. doi: 10.1021/jacs.7b10855
Q Zhou, K M Cobb, T Tan et al. J. Am. Chem. Soc., 2016, 138:12057~12060. doi: 10.1021/jacs.6b08075

图式 1 Ni催化的Suzuki偶联反应可能机理
Scheme 1 Possible mechanism of Ni-catalyzed Suzuki coupling reaction

图 1 C-C还原消除和氧化加成过程的能量(kcal/mol)
Figure 1 Energies of C-C reductive elimination and oxidative addition process(in kcal/mol)

图式 3 Ni催化甲基醚类底物的Suzuki偶联反应
Scheme 3 Ni catalyzed Suzuki coupling reaction of methyl ether substrates

图式 4 Ni催化苯基酯类底物的Suzuki偶联反应的可能机理
Scheme 4 Possible mechanism of Ni-catalyzed Suzuki coupling reaction for phenyl ester substrates

图 2 苯基酯类底物不同位置C-O键断裂的能量图
Figure 2 Energies of C-O bond cleaved at different positions of phenyl ester substrates

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

























 下载:
下载:

