表 1
CBP/P300抑制剂结构及活性
Table 1.
Structure and activity of CBP/P300 inhibitors

CBP和P300分别能够与环磷酸腺苷反应元件结合蛋白(CREB)和腺病毒早期区域1A蛋白(E1A)特异性结合,两者具有高度同源性,是许多转录因子的共活化剂[1~3]。CREBBP基因位于16号染色体p13.3,编码具有2442个氨基酸的CREBBP蛋白,EP300基因位于22号染色体q13.2,编码含有2415个氨基酸的P300蛋白[4]。CBP和P300是组蛋白乙酰化酶的两个亚型,在多种组织中广泛表达,并能够介导组蛋白乙酰化修饰过程,参与基因的表达调控,在肿瘤的发生发展中起重要作用[5]。由于CBP和P300属于同一类蛋白质,又具有相近的氨基酸排序和功能,所以常把它们写为CBP/P300。
CBP/P300都含有几个结构区域,包括能够乙酰化组蛋白和非组蛋白的组蛋白乙酰转移酶(histone acetyl transferase,HAT)结构域、蛋白激酶A磷酸化位点、谷氨酰胺富含区、EIA癌蛋白结合区、C-末端和结合乙酰化赖氨酸(KAc)的表观遗传阅读器溴结构域(BRD)[6, 7]。CBP/P300能够通过它们的溴结构域与染色质结合,并且一旦与染色质结合,该复合物将通过额外的转录机制来调节基因表达[8]。CBP/P300的溴结构域能够调节在肿瘤中广泛表达的转录因子和癌基因MYC,因此CBP/P300被认为是潜在的肿瘤靶点[9~12],其溴结构域也成为了一个新颖的治疗靶标,提供了基于溴结构域的靶向治疗恶性肿瘤新策略。为进一步了解CBP/P300的生物学特性和潜在疗法,研究人员已经发现了可以抑制CBP/P300溴结构域的各种化学探针[13~15]。
三维定量构效关系(3D-QSAR)被广泛用于药物分子结构与活性的研究,以此来设计新的药物。比较分子力场分析法(CoMFA)[16]和比较分子相似性指数分析法(CoMSIA)[17]是3D-QSAR的主要研究方法,这两种方法根据药物分子周围力场的差异,对药物活性进行回归分析得到受体与药物小分子配体之间的相互作用关系,进而选择性地设计活性更高的药物。本文采用CoMFA和CoMSIA方法研究CBP/P300溴结构域联芳基类抑制剂三维定量构效关系,即根据Lai等[18]合成的35个抑制剂分子建立3D-QSAR模型,确定联芳基类抑制剂分子结构与生物活性之间的定量关系。根据CoMFA和CoMSIA模型所提供的立体场、静电场、疏水场、氢键给体场、氢键供体场等信息提出改善此类抑制剂活性的药物设计思路,从而设计活性更高的新分子,为预测更加有效的CBP/P300溴结构域抑制剂提供理论依据。
Lai等[18]合成的35个CBP/P300溴结构域抑制剂分子结构如表 1所示,他们提供的化合物活性值为半数抑制浓度IC50,根据研究需要,需将该活性数值转换为pIC50值(-logIC50),列入表 1。
 下载:
导出CSV
下载:
导出CSV
| Compd. | Template Structure | R1 | R2 | Ar | R | pIC50 |
| 1 |  |
H |  |
- | - | 6.131 |
| 2 |  |
 |
- | - | 6.824 | |
| 3 |  |
 |
- | - | 6.620 | |
| 4* |  |
 |
- | - | 7.509 | |
| 5 |  |
- | - |  |
- | 6.921 |
| 6 | - | - |  |
- | 7.367 | |
| 7 | - | - |  |
- | 7.102 | |
| 8* | - | - |  |
- | 7.092 | |
| 9 | - | - |  |
- | 7.523 | |
| 10 | - | - |  |
- | 6.886 | |
| 11 | - | - |  |
- | 7.046 | |
| 12* | - | - |  |
- | 6.721 | |
| 13 |  |
- | - | - | 4-Cl | 7.377 |
| 14 | - | - | - | 4-Et | 7.420 | |
| 15 | - | - | - | 4-CH2OH | 7.092 | |
| 16* | - | - | - | 4-OMe | 7.638 | |
| 17 | - | - | - | 4-CN | 6.745 | |
| 18 | - | - | - | 5-Cl | 7.046 | |
| 19 | - | - | - | 5-Me | 7.886 | |
| 20* | - | - | - | 5-Et | 7.509 | |
| 21 | - | - | - | 5-CH2OH | 7.187 | |
| 22 | - | - | - | 5-OMe | 7.569 | |
| 23 | - | - | - | 5-CN | 6.553 | |
| 24* | - | - | - | 6-Cl | 6.770 | |
| 25 | - | - | - | 6-Me | 7.237 | |
| 26 | - | - | - | 6-Et | 7.620 | |
| 27 | - | - | - | 6-CH2OH | 7.161 | |
| 28* | - | - | - | 6-OMe | 7.161 | |
| 29 | - | - | - | 6-CN | 6.187 | |
| 30 |  |
- | - |  |
- | 8.921 |
| 31 | - | - |  |
- | 8.553 | |
| 32* | - | - |  |
- | 8.481 | |
| 33 | - | - |  |
- | 8.481 | |
| 34 | - | - |  |
- | 8.620 | |
| 35 | - | - |  |
- | 8.921 | |
| 注:*测试集分子 | ||||||
构建35个联芳基类抑制剂分子的三维结构,加氢处理后,通过最陡下降法(收敛RMS参数:0.05kcal ·mol-1 ·Å-1,最大迭代步数:6000,其他参数选择缺省值)和共轭梯度法(收敛RMS参数:0.001kcal ·mol-1 ·Å-1,最大迭代步数:1000,其他参数选择缺省值)优化分子构象。分子电荷的计算采用Gasteiger-Hückel法,最终得到能量最低的药物分子构象。
分子叠合效果良好是构建有效3D-QSAR模型的关键,3D-QSAR模型假定每个药物分子以同样的活性方式与受体的同一位点结合,所以选择活性最高的No.35分子(pIC50:8.921)作为模板进行基于骨架结构的分子叠合能够得到最好的叠合效果,计算程序为Align Database,使35个药物分子结构的共同部分叠合在同一刚性骨架上,并自动生成新的数据库。
CoMFA模型的构建是将分子叠合数据库中的药物分子置于软件设定的三维网格中,以sp3杂化的C+为探针,范德华半径为1.52Å[19],探针在三维网格中计算的步长设为2Å,计算和探测抑制剂分子周围的立体场(steric,S)和静电场(electrostaic,E),两种场效应能的阈值均设为30kcal·mol-1[20]。CoMSIA模型计算所采用的三维网格与CoMFA模型相同,并用相同的C+探针计算和预测药物分子周围的立体场、静电场、疏水场(hydrophobic,H)、氢键受体场(acceptor,A)和氢键给体场(donor,D),得到CBP/P300溴结构域联芳基类抑制剂分子结构与生物活性之间的定量关系。疏水特性为+1,氢键给体和氢键受体的强度均为+1。
本研究从35个抑制剂分子中随机选取27个分子作为训练集,采用偏最小二乘法结合pIC50活性数据与其相关的场描述符进行PLS分析得到CoMFA和CoMSIA模型,即采用抽一法(Leave-One-Out,LOO)对训练集药物分子进行交叉验证,得到最佳主成分数和交叉验证系数q2,调整柱滤值(column filtering value,σ)使得q2最大。再用非交叉验证法(No Validation)建立QSAR模型,得到非交叉验证系数r2、标准偏差SE和显著系数F,以此评估QSAR模型的预测能力。另外8个分子作为测试集检验模型的预测能力。以上计算均在SYBYL-X 2.0分子模拟软件中进行。
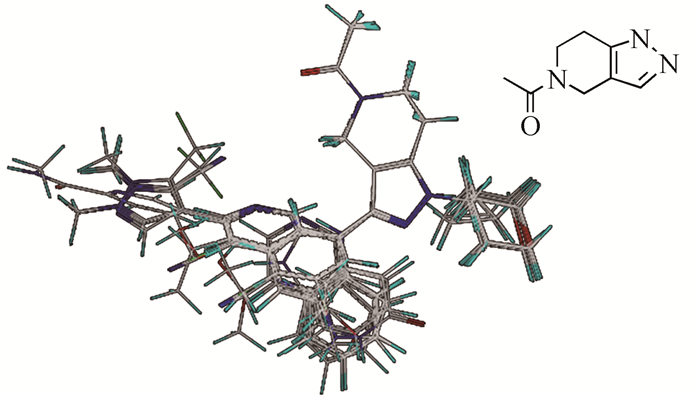
以活性最高的化合物35为模板,将35个CBP/P300联芳基类抑制剂分子按照基于公共骨架的方法进行叠合,叠合结果及叠合基本骨架如图 1所示,可以看到所有联芳基类抑制剂分子的公共骨架叠合在一起,相似基团的叠合取向也趋于一致,叠合效果良好。
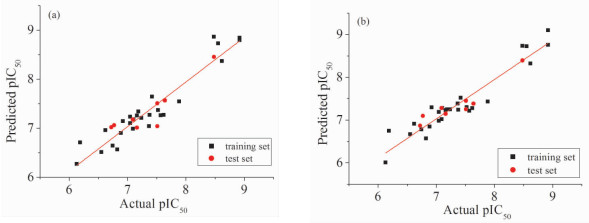
运用偏最小二乘法对训练集中27个抑制剂分子建立CoMFA模型,得到在最佳主成分数为3、柱滤值为5kJ/mol时交叉验证系数q2最大,为0.701。一般来讲,当q2大于0.5时认为模型具有良好的统计学意义[21]。非交叉验证系数r2=0.851,当r2大于0.6时,认为构建的模型具有较高的预测能力[22]。显著系数F=59.051,即所得数据具有较高的显著性。标准偏差SE=0.289,测试集中药物分子的pIC50真实值与预测值的偏差均小于1(表 2、表 3),说明我们建立的CoMFA模型的预测能力较为理想,pIC50真实值和预测值之间的线性关系见图 2(a)。
 下载:
导出CSV
下载:
导出CSV
| 3D-QSAR | ONC | q2 | SE | r2 | F | Field Contribution | |||||
| S | E | H | D | A | |||||||
| CoMFA | 3 | 0.701 | 0.289 | 0.851 | 59.051 | 0.538 | 0.462 | ||||
| CoMSIA | 4 | 0.629 | 0.230 | 0.909 | 74.868 | 0.193 | 0.326 | 0.091 | 0.225 | 0.166 | |
| 注:ONC:最佳主成分数 | |||||||||||
下载:
导出CSV
| Compd. | Actual pIC50 | CoMFA | CoMSIA | |||
| Predicted pIC50 | Residual | Predicted pIC50 | Residual | |||
| 4 | 7.509 | 7.042 | 0.467 | 7.252 | 0.257 | |
| 8 | 7.092 | 7.179 | -0.087 | 7.281 | -0.189 | |
| 12 | 6.721 | 7.020 | -0.299 | 6.869 | -0.148 | |
| 16 | 7.638 | 7.568 | 0.070 | 7.384 | 0.254 | |
| 20 | 7.509 | 7.508 | 0.001 | 7.451 | 0.058 | |
| 24 | 6.770 | 7.061 | -0.290 | 7.097 | -0.327 | |
| 28 | 7.161 | 7.012 | 0.149 | 7.146 | 0.015 | |
| 32 | 8.481 | 8.455 | 0.025 | 8.394 | 0.087 | |
| 35(template molecule) | 8.921 | 8.791 | 0.130 | 9.147 | -0.226 | |
| 注:模板分子35在训练集中pIC50预测值为CoMFA:8.738(Residual:0.183);CoMSIA:9.153(Residual:-0.232) | ||||||
运用偏最小二乘法对训练集中27个抑制剂分子建立CoMSIA模型,可知在最佳主成分数为4、柱滤值为3kJ/mol时得到的交叉验证系数最大,q2=0.629,非交叉验证系数r2=0.909,显著系数F=74.868,标准偏差SE=0.230,测试集中抑制剂分子的pIC50真实值与预测值之间的残差值均小于1,表明CoMSIA模型的预测能力也比较理想(表 2、3)。图 2(b)为CoMSIA模型中pIC50真实值和预测值的线性关系。
CoMFA模型中,立体场(S)的贡献率为53.8%,静电场(E)的贡献率为46. 2%,说明联芳基类抑制剂分子周围的立体场对其抑制CBP/P300溴结构域活性发挥主要作用。CoMSIA模型中立体场(S)的贡献率为19.3%,静电场(E)的贡献率为32.6%,疏水场(H)的贡献率为9.1%,氢键供体场(D)的贡献率为22.5%,氢键受体场(A)的贡献率为16.6%。根据CoMSIA模型提供的更多场信息,结合CoMFA模型共同提供了更加可靠的药物设计信息。
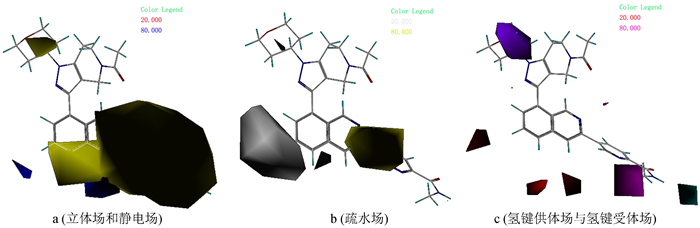
CoMFA模型等势图如图 3所示,以联芳基类抑制剂中活性最高的分子35作为参考分子进行分析。立体场区域用绿色和黄色表示,其中绿色代表增大基团有利于增加药物活性,黄色表示增大基团会降低药物活性,因此可在四氢吡喃或-Ar取代基位置引入-F、-Cl、-Br等体积较小的基团来提高药物分子活性。静电场区域用红色和蓝色表示,其中红色代表增加负电性基团有利于提高药物活性,蓝色代表增加正电荷基团有利于提高药物活性,因此可以在-Ar取代基位置引入-F、-Cl、-I等负电性基团,并在异喹啉5位原子引入正电荷基团如含氮官能团,来设计理论上药物活性更高的分子。
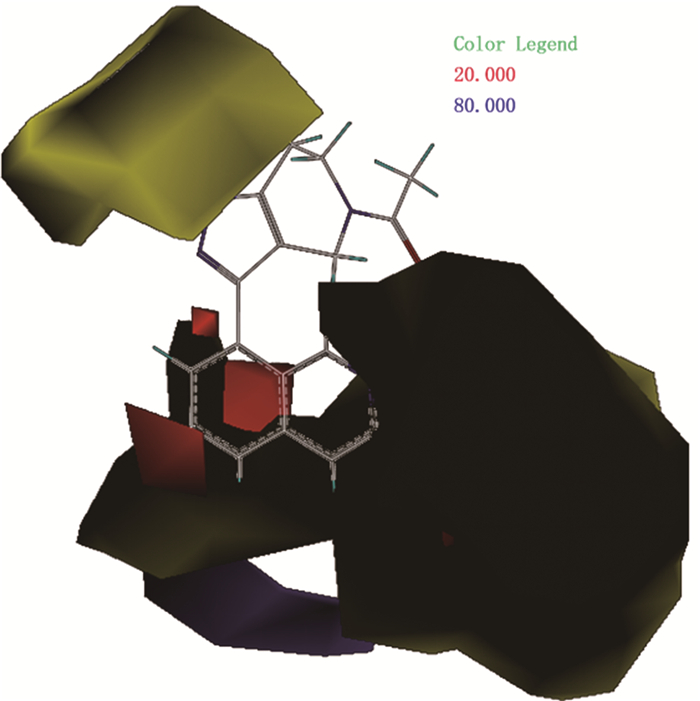
相比于CoMFA模型,CoMSIA模型提供了更多的药物分子设计信息,场贡献方面CoMFA模型只给出了立体场(S)与静电场(E),而CoMSIA模型除了提供立体场(S)和静电场(E)外,还有疏水场(H)、氢键受体场(A)和氢键给体场(D)。分析CoMSIA场贡献时,参考分子仍然选择pIC50值最大的抑制剂分子35。图 4(a)为CoMSIA模型立体场与静电场贡献图,场中颜色所代表的信息同CoMFA模型,可知在四氢吡喃与异喹啉取代基位置不宜引入较大基团,否则会降低药物活性,可以考虑引入-F、-Cl、-Br等体积较小的基团来提高药物分子活性。同时可在异喹啉5、6位上引入含氮基团等带正电基团提高药物活性。图 4(b)为CoMSIA模型疏水场贡献图,其中黄色代表增加疏水性基团有利于提高药物分子活性,白色代表增加亲水性基团有利于提高药物活性。黄色区域集中在-Ar处,说明在该处取代疏水性强的基团能够增加药物分子活性,如烷烃、环烷烃、芳香烃或酯基等基团; 而白色区域集中在异喹啉的5、6位,提示可以在该处引入羟基、氨基、羧酸基等亲水性基团来提高药物活性。图 4(c)为CoMSIA模型氢键受体场和氢键供体场贡献图,其中蓝绿色代表增加氢键供体有利于提高药物分子活性,紫色代表增加氢键供体不利于药物分子活性,紫红色代表增加氢键受体有利于提高药物活性,红色则表示增加氢键受体不利于药物活性。由此可知,可在-Ar中的-NH处引入氢键供体基团,如-OH等,并在其末端的甲基位置适当增加氢键受体,理论上可得到活性更高的CBP/P300抑制剂分子。
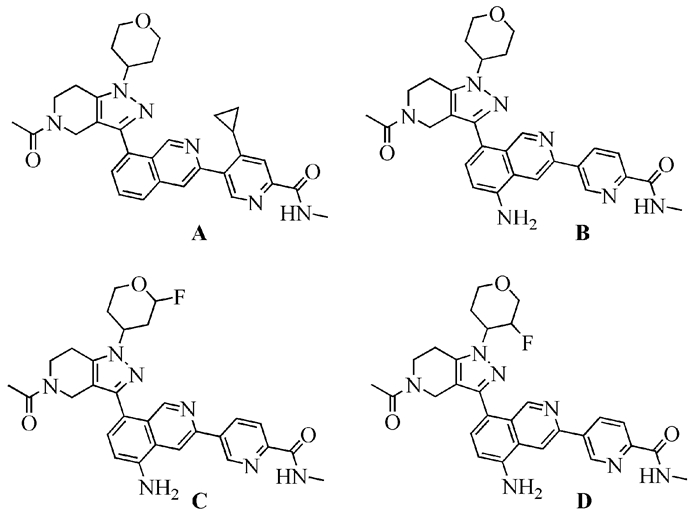
结合CoMFA与CoMSIA模型等势图提供的药物分子设计信息,我们设计了一系列新分子,并用CoMFA模型与CoMSIA模型预测这些新分子的pIC50值,最终得到四种理论上具有更高活性的化合物分子,其分子结构式见图 5,预测活性值见表 4。化合物A在-Ar处引入了环丙烷;化合物B在异喹啉5位处引入了-NH2;化合物C与D在异喹啉5位处引入了-NH2,并在四氢吡喃不同位置引入了-F。四个化合物经CoMFA模型与CoMSIA模型预测的pIC50值均高于模板分子35,提示这些新分子有望成为更加有效的CBP/P300抑制剂,这些新分子有待今后实验合成并确定其药效。
下载:
导出CSV
| Compd. | pIC50 (CoMFA) | pIC50 (CoMSIA) |
| A | 8.848 | 9.170 |
| B | 8.817 | 9.561 |
| C | 8.821 | 9.556 |
| D | 8.823 | 9.564 |
本研究对CBP/P300溴结构域联芳基类抑制剂进行了三维定量构效关系研究,采用偏最小二乘法建立了预测能力和统计学意义良好的CoMFA模型和CoMSIA模型,两模型训练集的交叉验证系数q2在0.6~0.7,非交叉验证系数r2在0. 8~0.9,标准偏差SE在0. 2~0.3。两模型活性数据pIC50的预测值与实验值基本一致,说明我们构建的CoMFA和CoMSIA模型可靠性非常高。结合3D-QSAR等势图中给出的立体场、静电场、疏水场、氢键供体场与氢键受体场分析了联芳基类抑制剂分子与受体间的相互作用情况。根据CoMFA等势图给出的药物设计信息可知,在四氢吡喃处引入-F、-Cl等体积较小的基团,在-Ar取代基位置引入-F、-Cl等体积较小且负电性高的基团,并在异喹啉5位原子引入含氮基团等带正电荷基团,能够提高药物分子活性。根据CoMSIA等势图可知,在四氢吡喃位置引入-F、-Cl等体积较小的基团,在异喹啉5、6位上引入体积较小的带正电基团或者羟基、氨基、羧酸基等亲水性基团能够增加药物活性。在-Ar位置取代烷烃、环烷烃、芳香烃等疏水性强的基团,在-Ar中的-NH处引入氢键供体,并在其末端的甲基位置适当增加氢键受体,可得到活性更高的抑制剂分子。CoMFA和CoMSIA模型共同提示的药物设计信息基本相符,基于此,我们设计了四种理论上具有更高活性的新分子,为预测更加有效的CBP/P300抑制剂提供可靠依据。
M Delvecchio, J Gaucher, C Aguilar-Gurrieri et al. Nat. Struct. Mol. Biol., 2013, 20: 1040-1046. doi: 10.1038/nsmb.2642
N Vo, R H Goodman. J. Biol. Chem., 2001, 276: 13505-13508. doi: 10.1074/jbc.R000025200
S Mujtaba, Y He, L Zeng et al. Mol. Cell, 2004, 13: 251-263. doi: 10.1016/S1097-2765(03)00528-8
J Wincent, A Luthman, M van Belzen et al. Mol. Genet. Genomic. Med., 2015, 4(1): 39-45.
蒋大贵, 刘玲娟, 吕铁伟等. 第三军医大学学报, 2016, 38(18): 2018-2022.
A Ito, C H Lai, X Zhao et al. EMBO J., 2001, 20: 1331-1340. doi: 10.1093/emboj/20.6.1331
E K Schorry, M Keddache, N Lanphear et al. Am. J. Med. Genet. A, 2008, 146A(19): 2512-2519.
I Radhakrishnan, G C Perez-Alvarado, D Parker et al. Cell, 1997, 91: 741-752. doi: 10.1016/S0092-8674(00)80463-8
L Jin, J Garcia, E Chan et al. Cancer Res., 2017, 77(20): 5564-5575. doi: 10.1158/0008-5472.CAN-17-0314
J H Kim, E J Cho, S T Kim et al. Nat. Struct. Mol. Biol., 2005, 12: 423-428. doi: 10.1038/nsmb924
J Vervoorts, J M Lüscherfirzlaff, S Rottmann et al. EMBO Rep., 2003, 4: 484-490. doi: 10.1038/sj.embor.embor821
A R Conery, R C Centore, A Neiss et al. eLife, 2016, e10483.
T D Crawford, F A Romero, K W Lai et al. J. Med. Chem., 2016, 59: 10549-10563. doi: 10.1021/acs.jmedchem.6b01022
P Filippakopoulos, O Fedorov, S Picaud et al. Angew. Chem. Int. Ed., 2014, 53: 6126-6130. doi: 10.1002/anie.201402750
S Picaud, O Fedorov, A Thanasopoulou et al. Cancer Res., 2015, 75: 5106-5119. doi: 10.1158/0008-5472.CAN-15-0236
R D Cramer, D E Patterson, J D Bunce. J. Am. Chem. Soc., 1988, 110: 5959-5967. doi: 10.1021/ja00226a005
G Klebe, U Abraham, T Mietzner. J. Med. Chem., 1994, 37: 4130-4146. doi: 10.1021/jm00050a010
K W Lai, F A Romero, V Tsui et al. Bioorg. Med. Chem. Lett., 2018, 28: 15-23. doi: 10.1016/j.bmcl.2017.11.025
时煜, 王小芳, 杨光中. 计算机与应用化学, 2002, 19(z1): 35-40.
杨伟华, 冯长君. 计算机与应用化学, 2007, 24(5): 688-692.
王新宇. 青岛科技大学硕士学位论文, 2011.
李国栋, 彭发, 陈兰美等. 化学研究与应用, 2015, 27(02): 113-121.

图 2 3D-QSAR模型pIC50预测值和实验值的线性关系: (a) CoMFA模型; (b) CoMSIA模型
Figure 2 Linear relationship between predicted pIC50 and actual pIC50 of 3D-QSAR Models: (a) CoMFA model; (b) CoMSIA model

图 3 CoMFA模型立体场和静电场贡献图
Figure 3 The contour map of CoMFA model for steric and electrostatic
表 1 CBP/P300抑制剂结构及活性
Table 1. Structure and activity of CBP/P300 inhibitors
| Compd. | Template Structure | R1 | R2 | Ar | R | pIC50 |
| 1 | |
H | |
- | - | 6.131 |
| 2 | |
|
- | - | 6.824 | |
| 3 | |
|
- | - | 6.620 | |
| 4* | |
|
- | - | 7.509 | |
| 5 | |
- | - | |
- | 6.921 |
| 6 | - | - | |
- | 7.367 | |
| 7 | - | - | |
- | 7.102 | |
| 8* | - | - | |
- | 7.092 | |
| 9 | - | - | |
- | 7.523 | |
| 10 | - | - | |
- | 6.886 | |
| 11 | - | - | |
- | 7.046 | |
| 12* | - | - | |
- | 6.721 | |
| 13 | |
- | - | - | 4-Cl | 7.377 |
| 14 | - | - | - | 4-Et | 7.420 | |
| 15 | - | - | - | 4-CH2OH | 7.092 | |
| 16* | - | - | - | 4-OMe | 7.638 | |
| 17 | - | - | - | 4-CN | 6.745 | |
| 18 | - | - | - | 5-Cl | 7.046 | |
| 19 | - | - | - | 5-Me | 7.886 | |
| 20* | - | - | - | 5-Et | 7.509 | |
| 21 | - | - | - | 5-CH2OH | 7.187 | |
| 22 | - | - | - | 5-OMe | 7.569 | |
| 23 | - | - | - | 5-CN | 6.553 | |
| 24* | - | - | - | 6-Cl | 6.770 | |
| 25 | - | - | - | 6-Me | 7.237 | |
| 26 | - | - | - | 6-Et | 7.620 | |
| 27 | - | - | - | 6-CH2OH | 7.161 | |
| 28* | - | - | - | 6-OMe | 7.161 | |
| 29 | - | - | - | 6-CN | 6.187 | |
| 30 | |
- | - | |
- | 8.921 |
| 31 | - | - | |
- | 8.553 | |
| 32* | - | - | |
- | 8.481 | |
| 33 | - | - | |
- | 8.481 | |
| 34 | - | - | |
- | 8.620 | |
| 35 | - | - | |
- | 8.921 | |
| 注:*测试集分子 | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 CoMFA和CoMSIA模型数据参数
Table 2. Statistical parameters of CoMFA and CoMSIA models of training set
| 3D-QSAR | ONC | q2 | SE | r2 | F | Field Contribution | |||||
| S | E | H | D | A | |||||||
| CoMFA | 3 | 0.701 | 0.289 | 0.851 | 59.051 | 0.538 | 0.462 | ||||
| CoMSIA | 4 | 0.629 | 0.230 | 0.909 | 74.868 | 0.193 | 0.326 | 0.091 | 0.225 | 0.166 | |
| 注:ONC:最佳主成分数 | |||||||||||
下载: 导出CSV
表 3 CoMFA和CoMSIA模型测试集pIC50预测值与真实值比较
Table 3. Comparison of predicted and actual pIC50 of test set for the CoMFA and CoMSIA models
| Compd. | Actual pIC50 | CoMFA | CoMSIA | |||
| Predicted pIC50 | Residual | Predicted pIC50 | Residual | |||
| 4 | 7.509 | 7.042 | 0.467 | 7.252 | 0.257 | |
| 8 | 7.092 | 7.179 | -0.087 | 7.281 | -0.189 | |
| 12 | 6.721 | 7.020 | -0.299 | 6.869 | -0.148 | |
| 16 | 7.638 | 7.568 | 0.070 | 7.384 | 0.254 | |
| 20 | 7.509 | 7.508 | 0.001 | 7.451 | 0.058 | |
| 24 | 6.770 | 7.061 | -0.290 | 7.097 | -0.327 | |
| 28 | 7.161 | 7.012 | 0.149 | 7.146 | 0.015 | |
| 32 | 8.481 | 8.455 | 0.025 | 8.394 | 0.087 | |
| 35(template molecule) | 8.921 | 8.791 | 0.130 | 9.147 | -0.226 | |
| 注:模板分子35在训练集中pIC50预测值为CoMFA:8.738(Residual:0.183);CoMSIA:9.153(Residual:-0.232) | ||||||
下载: 导出CSV
表 4 CoMFA和CoMSIA模型预测新分子的pIC50值
Table 4. The predicted pIC50 of novel molecules for the CoMFA and CoMSIA models
| Compd. | pIC50 (CoMFA) | pIC50 (CoMSIA) |
| A | 8.848 | 9.170 |
| B | 8.817 | 9.561 |
| C | 8.821 | 9.556 |
| D | 8.823 | 9.564 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们