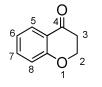
 图 图式1
4-色满酮的骨架结构
Figure 图式1.
Structure of 4-chromanone
图 图式1
4-色满酮的骨架结构
Figure 图式1.
Structure of 4-chromanone
引用本文:
王桂霞, 张洲洋, 刘鹏, 廖培海, 孔翔飞. 具有生物活性的4-色满酮衍生物的合成研究进展[J]. 化学通报,
2017, 80(4): 322-328.

Citation: Wang Guixia, Zhang Zhouyang, Liu Peng, Liao Peihai, Kong Xiangfei. Progress in the Syntheses of 4-Chromanone Derivatives with Bioactivities[J]. Chemistry, 2017, 80(4): 322-328.
Citation: Wang Guixia, Zhang Zhouyang, Liu Peng, Liao Peihai, Kong Xiangfei. Progress in the Syntheses of 4-Chromanone Derivatives with Bioactivities[J]. Chemistry, 2017, 80(4): 322-328.
具有生物活性的4-色满酮衍生物的合成研究进展
English
Progress in the Syntheses of 4-Chromanone Derivatives with Bioactivities
Abstract:
4-Chromanone derivatives are widespread in nature, most of them have biological activities. 4-Chromanone derivatives with bioactivities are chiral compounds, especially C2. The asymmetric synthesis of these compounds has attracted great attention. The key step of synthesizing of 4-chromanone derivatives is to control the synthesis of C2 chiral center. In this paper, based on the different synthesis routes, various methods of synthesizing C2 (racemic or chiral)-4-chromanone derivatives in recent years were summarized, and the reaction characteristics of synthesizing C2 (chiral)-4-chromanone derivatives were discussed.
-
Key words:
- 4-Chromanone derivatives
- / Bioactivity
- / Asymmetric synthesis
- / Cyclization
-
自从发现来源于自然界的4-色满酮类衍生物具有显著的抗炎活性[1]以来,有关此类衍生物的研究受到国内外有机化学家及药物化学家的极大关注。至今,文献报道的具有生物活性色满酮类衍生物总数已达上万个。学术界对4-色满酮类衍生物(其骨架结构见图式 1) 简单分成了两大类:C2-取代基中含有苯环的衍生物称为黄酮类衍生物,其他均称为4-色满酮类化合物,本文为了统一均称为4-色满酮类衍生物。
图 图式1
4-色满酮的骨架结构
Figure 图式1.
Structure of 4-chromanone
随着人们对其认识的不断深入,发现具有生物活性的4-色满酮类衍生物大多具有手性中心,尤其在它们的C2位置,如图式 2所示。化合物1:松属素,主要存在于蜂胶中,具有麻醉止痛的功效;化合物2:杜鹃素,主要存在杜鹃属植物中,具有祛痰功效;化合物3:球松甲素,主要存在于球松类植物中,具有抗动脉粥样化的功效;化合物4:2-氢汉黄芩素,主要从黄芩或夹竹桃等植物中获得,具有抗癌、利尿作用;化合物5:甘草黄素,由甘草中提取得到,具有解痉、抗菌等功效;化合物6:乔松酮,由山核桃的壳中提取得到,具有抗氧化、抗白血病的生物活性[2, 28];化合物7:橙皮素,柑橘属植物果实中的主要药效成分,有祛痰、镇咳等功效[3];化合物8:植物内生真菌中提取得到的4-色满酮衍生物,具有高效的抗菌效果[4];化合物9:从源于非洲的豆科植物中提取得到,具有抗结核功效[5];化合物10和11分别具有抗葡萄球菌和抗大肠杆菌的功效[6];化合物12是蛋白酶SIRT2的抑制剂,具有延缓人类衰老的功效[7]。有关生物活性C2(消旋或手性)-4-色满酮衍生物的文献报道层出不穷[8]。
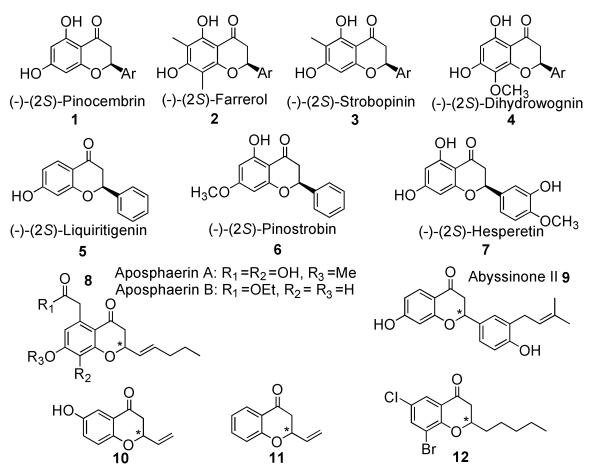 图 图式2
C2(消旋或手性)-4-色满酮衍生物
Figure 图式2.
C2 (racemic or chiral)-4-chromanone derivatives
图 图式2
C2(消旋或手性)-4-色满酮衍生物
Figure 图式2.
C2 (racemic or chiral)-4-chromanone derivatives
另外,C2(消旋或手性)-4-色满酮衍生物作为重要中间体,也是合成很多其他具有生物活性天然产物的前体[9]。例如,中药丹参中的活性成分紫草酸((+)lithospermic acid)(图式 3)[10]具有抗HIV病毒的生物活性,其合成的关键在于前体光学纯C2(手性)-4-色满酮衍生物的合成。红厚壳属植物中的提取物(+)-Calanolide A也具有抗HIV病毒活性,其合成前体也为C2(手性)-4-色满酮衍生物(图式 4)[11]。
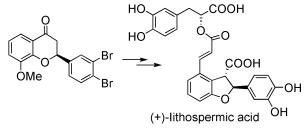 图 图式3
借助C2(手性)-4-色满酮衍生物合成紫草酸
Figure 图式3.
Synthesis of (+) lithospermic acid via C2 (chiral)-4-chromanone derivatives
图 图式3
借助C2(手性)-4-色满酮衍生物合成紫草酸
Figure 图式3.
Synthesis of (+) lithospermic acid via C2 (chiral)-4-chromanone derivatives
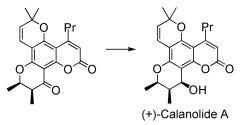 图 图式4
借助C2(手性)-4-色满酮衍生物合成(+)-Calanolide
Figure 图式4.
Synthesis of (+)-Calanolide via C2 (chiral)-4-chromanone derivatives
图 图式4
借助C2(手性)-4-色满酮衍生物合成(+)-Calanolide
Figure 图式4.
Synthesis of (+)-Calanolide via C2 (chiral)-4-chromanone derivatives
综上所述,C2(消旋或手性)-4-色满酮衍生物是一类非常重要的化合物。本文从合成方法学的角度,对近几十年来较典型的合成C2(消旋或手性)-4-色满酮衍生物的方法进行了归纳和总结,并侧重讨论了不对称合成立体结构专一的C2(手性)-4-色满酮衍生物的方法特点。
1 C2(消旋)-4-色满酮衍生物的合成
最初,科研工作者大多致力于C2(消旋)-4-色满酮衍生物的合成。1978年,Kabbe[12]首次报道了邻羟基苯乙酮与烷基酮或脂肪醛在四氢吡咯作用下构建C2(消旋)-4-色满酮衍生物(式(1))。此种方法反应底物范围广,产品产率高。很长一段时间内此方法成为合成工作者构建C2(消旋)-4-色满酮环的必经之路。该方法存在的主要缺陷是,反应物如果含有对碱敏感的基团,则目标产物的产率比较低或者根本不反应。
在这样的背景下,1991年,Kelly等[13]报道了酸性条件下合成C2(消旋)-4-色满酮衍生物的方法(式(2))。首先,底物邻羟基苯乙酮在三甲基氯化硅(TMSCl)和二异丙基氨基锂(LDA)的作用下得到中间体二烯醇硅醚,二烯醇硅醚在Lewis酸(TiCl4)作用下与酮加成并分子内成环得C2(消旋)-4-色满酮衍生物。此种方法为含有对碱性敏感基团的反应底物提供了一条构建C2(消旋)-4-色满酮衍生物的有效途径。
2005年,Chandrasekhar等[14]以邻羟基苯乙酮和醛(大部分为芳醛)为底物、L-脯氨酸为催化剂,合成了一系列C2(消旋)-4-色满酮衍生物(式(3))。这种合成方法反应条件温和,产率较高,但产物没有立体选择性。此外,Luthman等[15]还借助微波辐射合成了C2(消旋)-4-色满酮衍生物(式(4)),产率为43%~88%,该方法同样也不具备立体选择性。
2010年,Sharma等[16]报道了利用Julia-Kocienski烯化方法合成C2(消旋)-4-色满酮衍生物,反应底物为事先设计合成的Julia偶合试剂,在碱性条件下与芳醛经历烯化、分子内环化,成功得到C2(消旋)-4-色满酮衍生物,反应产率60%~70%。该报道的新颖之处在于Julia偶合试剂的利用,但此反应底物仅限于芳香醛,脂肪醛不适用此反应。
2011年,Chen等[17]报道了邻羟基苯乙酮与芳醛经Claisen-Schmidt和Mitsunobu反应得到C2(消旋)-4-色满酮衍生物;另外,使用同样的原料,以三乙烯二胺作为碱性试剂、水为溶剂,加热回流20h,可“一锅法”直接得到C2(消旋)-4-色满酮衍生物,产率67%~78%。此反应的创新点在于采用绿色环保的溶剂体系。
Kavala等[18]报道了在甲醇溶液中碘作为催化剂催化芳醛、苯胺、2-羟基苯乙酮三组分Mannich反应,“一锅法”得到C2(消旋)-4-色满酮衍生物,产率最高可达82%。分子碘作为Lewis酸表现出高的区域选择性和化学选择性,并且,在稀溶液、浓溶液或无溶剂情况下均表现出较高的反应活性。
Li等[19]报道了天然产物Gonytolide的合成。其中色满酮内脂单元的合成采用分子内的氧杂Michael加成反应,产率可高达99%,得到消旋的色满酮内脂,如式(8) 所示。采用手性色谱柱进行分离,可得到光学纯的目标产物。
以上是一些最具有代表性的C2(消旋)-4-色满酮衍生物合成的报道。近年来对光学活性的C2(手性)-4-色满酮衍生物需求逐渐增加,因此C2(手性)-4-色满酮衍生物的不对称合成得到迅速发展。
2 C2(手性)-4-色满酮衍生物的合成
自19世纪Fischer开创不对称合成领域以来,不对称反应技术主要经历了四个阶段:(1) 手性源的不对称反应;(2) 手性助剂的不对称反应;(3) 手性试剂的不对称反应;(4) 不对称催化反应。有关C2(手性)-4-色满酮衍生物的不对称合成的报道大致也经历了这样一个过程。
最初,C2(手性)-4-色满酮骨架结构是借助自然界中的生物酶催化来合成,其中最具有代表性的为查尔酮酶(Chalcone isomerase,简写CHI)[20],这种酶催化反应的立体选择性很高,接近100%,如式(9) 所示。
2001年,Hodgetts[21]通过手性源——卤代醇与邻溴基苯酚反应,构建了C2(手性)-4-色满衍生物,然后再氧化C4得到C2(手性)-4-色满酮衍生物,如式(10) 所示。此种合成方法中C2(手性)来源于手性卤代醇。
同期,也有科学家借助手性辅剂合成C2(手性)-4-色满酮衍生物,其中手性亚砜为最常用的手性辅剂之一[22]。Solladié等[23]借助手性亚砜辅剂,利用Kabbe缩合环化方法得到差向异构体C2(手性)-4-色满酮衍生物,采用普通方法分离得到C2立体结构专一的色满酮衍生物,然后再脱除色满酮衍生物C3位的手性亚砜,得到C2(手性)-4-色满酮衍生物(式(11))。
2003年,Kawasaki等[24]利用手性酶催化C2(消旋)端羧基酚醚与1-丁醇进行酯化反应(如式(12) 所示),结果1-丁醇仅与其中一种S构型酚醚发生酯化反应,从而实现对映体的拆分。拆分后的化合物分别缩合环化,得到构型分别为S和R的C2(手性)-4-色满酮衍生物。
借助手性源、手性辅剂等方法构建C2(手性)-4-色满酮衍生物总存在不尽人意之处,例如,合成路线长、产率低、原子经济性差;手性生物酶催化的反应,立体选择性高,产率高,但其主要缺陷在于酶催化反应的专一性及酶的易变性失活等。鉴于此,科学家将目光投向了不对称催化合成。不对称催化合成一般指利用合理设计的手性催化剂作为手性模板控制反应,将大量潜手性底物选择性地转化成特定构型的产物,实现手性放大和手性增值。不对称催化合成法已发展成为合成立体结构专一的手性化合物的最为经济有效方法。因此,有关不对称催化合成C2(手性)-4-色满酮衍生物的报道也相继出现。
2007年,Scheidt等[25]采用手性硫脲作催化剂,酚羟基与不饱和的β-酮酯分子内共轭加成成环,环化产物在对甲苯磺酸作用下脱去C3位取代基叔丁酯,得到C2(手性)-4-色满酮衍生物(式(13))。此种催化剂催化的反应条件温和、立体选择性高,对映体过量(ee)最高可达94%。
2008年,冯小明课题组[26]采用手性双氮氧-镍配合物催化分子内酚羟基与不饱和β-酮酯共轭加成成环,如式(14) 所示。反应底物及反应过程与Scheidt等[25]的方法相似,差别在于其手性催化剂不同。手性双氮氧-镍配合物催化的反应产率高、立体选择性高,ee值最高达99%。
以上两种不对称催化方法的共同特点为,反应的底物邻羟基不饱和β-酮酯化合物是由Knoevenagel缩合反应制备,且反应产物需通过C3-叔丁基酯基团的脱除得到。这在一定程度上降低了通过此方法构建C2(手性)-4-色满酮衍生物的竞争性。
利用查尔酮衍生物作为底物,经催化环化制备C2(手性)-4-色满酮衍生物更切实可行。Hintermann等[27]利用天然手性催化剂奎宁(QN)及对映体奎宁丁(QD)、辛可宁(CN)及对映体辛可宁丁(CD)作催化剂,催化底物邻羟基α, β-烯酮(查尔酮衍生物),一步法加成环化合成C2(手性)-4-色满酮衍生物,立体选择性最高达64%,如式(15) 所示。其中不对称催化效果最佳的为含有甲氧基的奎宁,可能为甲氧基基团的空间立体效应诱导而致。
Zhang等[31]在甲苯溶液中采用助剂4-氯苯甲酸和L-脯氨酸衍生物共同作用下催化2-羟基查尔酮环化,得到立体结构唯一的C2(手性)-4-色满酮衍生物,如式(16) 所示。他们考察了一系列L-脯氨酸衍生物的催化效果,其中立体选择性最佳的催化剂为3a。作者推测其较高的催化效果主要源于反应底物与催化剂π-π堆积的诱导作用。
2015年,Liu等[32]报道了三甲基碘硅烷(TMSI)诱导非对映选择性Michael加成反应,立体选择性最高可达90%(式(17))。这一方法为合成含有此结构的天然产物提供了便利的途径。
Liao等[28]利用铑与手性亚砜的配合物作催化剂,催化四芳基硼酸钠与4-色烯酮的双键加成,得到C2(手性)-4-色满酮衍生物(式(18)),立体选择性达99%。Korenaga等[29]在甲苯和水的混合溶剂中,利用铑与联苯形成的配合物为催化剂,不对称催化还原4-色烯酮得到C2(手性)-4-色满酮衍生物(式(19)),立体选择性可达99%。此类反应立体选择性好,但是反应原子经济性差,副产物有一定毒性,不利于药物中间体的合成。
2013年,Lemke等[30]采用甲酸/三乙胺混合溶剂,以金属配合物为催化剂,对C2(消旋)-4-色满酮衍生物的羰基进行不对称还原,其中一异构体还原得到(2R, 4R)产物,另一异构体被拆分得到光学纯化合物,拆分的立体选择性最高可达99%。他们筛选了一系列金属配合物,拆分效果最好的为含有2个苯基取代基的铑配合物Ⅲ,如式(20) 所示。
Vijayan等[33]采用二氯(五甲基环戊二烯基)合铑(Ⅲ)二聚体(缩写:[RhCl2Cp*]2)与Cu(OAc)2·H2O共同催化氧化水杨醛与杂多环烯烃稠化合环得到三环色满酮衍生物,产率最高可达70%。反应中,铑与水杨醛形成苯并五元环的过渡态,双键插入到五元环中,最终得到立体结构专一的三环色满酮衍生物(如式(21) 所示)。
以上代表性的C2(手性)-4-色满酮衍生物合成的报道为构建此类化合物提供了高效的合成方法,有关此方面的最新研究进展还可参见文献[34]。
3 结语
C2(消旋或手性)-4-色满酮衍生物是一类非常重要的化合物,有关它们的合成在有机合成领域非常引人注目,研究人员开发了不少C2(手性)-4-色满酮衍生物的高效不对称合成方法,为后续工作积累了宝贵经验。但是发展至今,有关C2(手性)-4-色满酮衍生物不对称合成仍有许多问题有待解决:首先,根据目前的文献报道,不对称催化合成的催化剂大部分是实验室设计并自行制备的有机金属催化剂或有机配合物催化剂,此类催化剂制备繁琐,底物适用范围窄,回收使用困难;其次,大部分用于不对称催化合成的催化剂虽然实现了手性增值,但是对环境不同程度的产生了负面影响,尤其是有机金属催化剂痕量重金属的存在制约了其在制药工业界的应用;再有,不对称催化反应底物比较有限,大部分不对称催化合成的报道的底物仅限于分子内的酚羟基与邻位基团的加成环化或4-色烯酮加氢。总而言之,有关C2(手性)-4-色满酮衍生物不对称合成仍有很多工作需要我们科研工作者继续完善。
-
-
[1]
(a) G P Ellis, The Chemistry of Heterocyclic Compounds. Wiley: New York, 1977, 31; (b) T W Wallace, Nat. Prod. Rep. , 1986, 465~476; (c)李慧嫒, 胡春. 中国药学杂志, 2005, 40 (4): 241~244; (d)郑伟, 孙光, 孙宝佳等. 精细化工中间体, 2009, 39(1): 30~33; (e) A E Nibbs, K A Scheidt. Eur. J. Org. Chem. , 2012, 449~462.
-
[2]
S Ramadas, G L Krupadanam. Tetrahedron: Asym. 2004, 15: 3381~3391. https://www.researchgate.net/publication/244241104_Enantioselective_acylation_of_()-_cis-flavan-4-ols_catalyzed_by_lipase_from_Candida_cylindracea_(_CCL)_and_the_synthesis_of_enantiopure_flavan-4-ones
-
[3]
(a) T Korenaga, K Hayashi, T Sakai et al. Org. Lett., 2011, 13: 2022~2025; (b) D Q Sun, G Julian. Chem. Med. Chem., 2012, 7: 1541~1545.
-
[4]
U Albrecht, M Lalk, P Langer. Bioorg. Med. Chem., 2005, 13: 1531~1536. https://www.researchgate.net/publication/8033464_Synthesis_and_structure-activity_relationships_of_2-vinylchroman-4-ones_as_potent_antibiotic_agents?ev=sim_pub
-
[5]
(a) D Sun, J G Hurdle, R Lee et al. Chem. Med. Chem., 2012, 7(9): 1541~1545; (b) V S Kamat, F Y Chuo, K Nakanishi. Heterocycles, 1981, 15: 1163~1170.
-
[6]
R S Keri, S Budagumpi, R K Pai. Eur. J. Med. Chem., 2014, 78: 340~374. https://www.researchgate.net/profile/Srinivasa_Budagumpi2/publication/261292063_Chromones_as_a_privileged_scaffold_in_drug_discovery_A_review/links/53f571d80cf2888a7491c0fe/Chromones-as-a-privileged-scaffold-in-drug-discovery-A-review.pdf
-
[7]
M Friden-Saxin, T Seifert, K Luthman. J. Med. Chem., 2012, 55: 7104~7113.
-
[8]
(a) H M Merken, G R Beecher et al. J. Agric. Food Chem., 2000, 48: 577~599; (b) H Y Chen, K D Dykstra, E T Birzin et al. Bioorg. Med. Chem. Lett., 2004, 14: 1417~1421; (c) M A Terzidis, J Stephanidou, C A Tsoleridis. J. Org. Chem., 2010, 75: 1948~1955; (d) N X Wang, Y L Xing, Y J Wang. Curr. Org. Chem., 2013, 17 (14): 1555~1562; (e) L Feng, M M Maddox, M Z Alam et al. J. Med. Chem., 2014, 57: 8398~8420.
-
[9]
(a) M E Bellizzi, A V Bhatia, C C Steven et al. Org. Proce. Res. Dev., 2014, 18: 303~309; (b) S Ghosh, N B Chandar. Synlett, 2014, 25: 2649~2653; (c) T Rosenau, A Potthast. Org. Lett., 2002, 4(8): 1257~1258.
-
[10]
A K Ghosh, X Cheng, B Zhou. Org. Lett., 2012, 14(19): 5046~5049.
-
[11]
E Sekin, T Kumamoto, T Ishikawa et al. J. Org. Chem., 2004, 69: 2760~2767.
-
[12]
(a) H L Kabbe. Synthesis, 1978, 12: 886~887; (b) H L Kabbe, A Widdig. Angew. Chem. Int. Ed., 1982, 21: 247~256.
-
[13]
S E Kelly, B C Vandeplas. J. Org. Chem., 1991, 56(3): 1325~1327.
-
[14]
S Chandrasekhar, K Vijeender, K V Reddy. Tetrahed. Lett., 2005, 46: 6991~6993. https://www.researchgate.net/publication/238461145_New_Synthesis_of_Flavanones_Catalyzed_by_L-Proline
-
[15]
(a) E Wallen, K Dahlen, M Grotli et al. Org. Lett., 2007, 9: 389~391; (b) F S Maria, N Pemberton, K Luthman et al. J. Org. Chem., 2009, 74: 2755~2759.
-
[16]
S K Sharma, V D Tripathi. Tetrahedron, 2010, 66: 9445~9449. https://www.researchgate.net/publication/229116571_Synthesis_of_chalcones_and_flavanones_using_JuliaKocienski_olefination
-
[17]
P Y Chen, T P Wang, M Y Chiang et al. Tetrahedron, 2011, 67: 4155~4164. https://www.researchgate.net/publication/251498560_Environmentally_benign_syntheses_of_flavanones
-
[18]
(a) V Kavala, C Lin, C Yao et al. Tetrahedron, 2012, 68: 1321~1329. (b) Y M Ren, C Cai, R C Yang. RSC Adv., 2013, 3: 7182~7204.
-
[19]
(a) F F Li, D J Atkinson, D P Furkert et al. Eur. J. Org. Chem., 2016, 1145~1155; (b) L F Tietze, S Jackenkroll, J Hierold. Chem. Eur. J., 2014, 20: 8628~8635; (c) T Qin, R P Johnson, J A Porco Jr. J. Am. Chem. Soc., 2011, 133: 1714~1717.
-
[20]
R A Bednar, J R Hadcock. J. Biol. Chem., 1988, 263: 9582~9588. https://www.researchgate.net/publication/19771149_Purification_and_characterization_of_chalcone_isomerase_from_soybeans
-
[21]
K J Hodgetts. Tetrahed. Lett., 2001, 42(22): 3763~3766.
-
[22]
李树坤, 白东鲁. 化学进展, 2001, 13(5): 351~358. http://www.jxndxuebao.com/index.php/HXJZ/article/view/1585051
-
[23]
(a) G Solladié, N Gehrold, J Maignan. Tetrahedron: Asymm., 1999, 10: 2739~2747; (b) S T Saengchantara, T W Wallace. Tetrahedron, 1990, 46: 6553~6564.
-
[24]
M Kawasaki, H Kakuda, M Goto. Tetrahedron: Asym., 2003, 14(11): 1529~1534. M Kawasaki, H Kakuda, M Goto. Tetrahedron: Asym., 2003, 14(11): 1529~1534.
-
[25]
(a) M M Biddle, M Lin, K A Scheidt. J. Am. Chem. Soc., 2007, 129: 3830~3831; (b) A E Nibbs, K A Scheidt. Eur. J. Org. Chem., 2012, 3: 449~462.
-
[26]
(a) L J Wang, X H Liu, X M Feng et al. Angew. Chem., 2008, 120: 8798~8801; (b) L J Wang, X H Liu, X M Feng et al. Angew. Chem. Int. Ed., 2008, 47: 8670~8673.
-
[27]
(a) C Dittmer, G Raabe, L Hintermann. Eur. J. Org. Chem., 2007, 5886~5898; (b) L Hintermann, C Dittmer. Eur. J. Org. Chem., 2012, 5573~5584.
-
[28]
(a) J Chen, J M Chen, J Liao et al. J. Am. Chem. Soc.. 2010, 132(13) 4552~4553; (b) F Han, G Chen, J Liao et al. Eur. J. Org. Chem. 2011, 2928~2931.
-
[29]
T Korenaga, K Hayashi, Y Akaki et al. Org. Lett., 2011, 13(8), 2022~2025. https://www.researchgate.net/publication/50418507_Highly_enantioselective_and_efficient_synthesis_of_flavanones_including_pinostrobin_through_the_rhodium-catalyzed_asymmetric_14-addition
-
[30]
M K Lemke, S Pia, F Petra et al. Angew. Chem. Int. Ed. 2013, 52, 11651~11655. https://www.researchgate.net/publication/262855856_Ein_praktischer_Zugang_zu_hoch_enantiomerenreinen_Flavanonen_durch_katalytische_asymmetrische_Transferhydrierung
-
[31]
Y L Zhang, Y Q Wang. Tetrahed. Lett., 2014, 55: 3255~3258.
-
[32]
J Liu, Z. Li, P Tong et al. J. Org. Chem., 2015, 80: 1632~1643. https://www.researchgate.net/publication/270652028_TMSI-Promoted_Vinylogous_Michael_Addition_of_Siloxyfuran_to_2-Substituted_Chromones_A_General_Approach_for_the_Total_Synthesis_of_Chromanone_Lactone_Natural_Products
-
[33]
(a) A Vijayan, T V Baiju, E Jijy et al. Tetrahedron, 2016, 72: 4007~4015; (b) E Jijy, P Prakash, M Shimi et al. Chem. Commun., 2013, 49: 7349~7351.
-
[34]
S Emamia, Z Ghanbarimasir. Eur. J. Med. Chem., 2015, 93: 539~563. https://www.researchgate.net/publication/273147375_Recent_advances_of_chroman-4-one_derivatives_Synthetic_approaches_and_bioactivities
-
[1]
-

图式3 借助C2(手性)-4-色满酮衍生物合成紫草酸
Scheme 3 Synthesis of (+) lithospermic acid via C2 (chiral)-4-chromanone derivatives
-


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 10
- 文章访问数: 1672
- HTML全文浏览量: 527
 下载:
下载:
