 图1
废弃SCR脱硝催化剂
Figure1.
Waste SCR denitrification catalyst
图1
废弃SCR脱硝催化剂
Figure1.
Waste SCR denitrification catalyst
引用本文:
唐昊, 陆强, 杨江毅, 李慧, 李文艳, 杨勇平. 废弃SCR催化剂的循环再利用及表征分析研究[J]. 燃料化学学报,
2018, 46(2): 233-242.

Citation: TANG Hao, LU Qiang, YANG Jiang-yi, LI Hui, LI Wen-yan, YANG Yong-ping. Research on recycling and characterization analysis of the waste SCR catalyst[J]. Journal of Fuel Chemistry and Technology, 2018, 46(2): 233-242.
Citation: TANG Hao, LU Qiang, YANG Jiang-yi, LI Hui, LI Wen-yan, YANG Yong-ping. Research on recycling and characterization analysis of the waste SCR catalyst[J]. Journal of Fuel Chemistry and Technology, 2018, 46(2): 233-242.
废弃SCR催化剂的循环再利用及表征分析研究
摘要:
针对废弃SCR脱硝催化剂常规再利用处理后存在SO2氧化率高的问题, 提出了一种新型的废弃催化剂再利用新工艺, 包括酸洗、还原酸浸和活性组分负载等步骤, 以有效控制再利用催化剂的SO2氧化率。实验考察了经不同步骤处理后所得催化剂的组分、脱硝效率和SO2氧化率的变化情况, 并对催化剂进行了深入的分析表征。结果表明, 新鲜催化剂、废弃催化剂、常规再利用催化剂、新型再利用催化剂的脱硝效率和SO2氧化率分别为99.0%和0.43%、77.0%和0.46%、94.2%和0.80%、99.3%和0.48%, 采用本方法获得的再利用催化剂不仅脱硝效率完全恢复, 而且SO2氧化率得到了很好的控制。通过对催化剂的分析表征发现, 采用常规再利用技术不能有效清除废弃催化剂表面的高聚态钒物种, 而本方法则可以有效清理这类高聚态钒物种, 并以高度分散的钒物种进行替代, 从而有效控制再利用催化剂的SO2氧化率。
English
Research on recycling and characterization analysis of the waste SCR catalyst
Abstract:
In the light of a high SO2 oxidation ratio of the recycled waste SCR catalyst during conventional recycling denitrification, a novel recycling technique, including acid washing, acid leaching under reduction condition and active components loading, was proposed to control the SO2 oxidation ratio of the recycled catalyst.The experiments were performed to determine the components, denitrification efficiency and SO2 oxidation ratio of the catalysts obtained in different recycling runs, and the catalysts used were characterized.The results indicate that the denitrification efficiency and SO2 oxidation ratio of the fresh catalyst, waste catalyst, conventional recycled catalyst and novel recycled catalyst are 99.0% and 0.43%, 77.0% and 0.46%, 94.2% and 0.80%, 99.3% and 0.48%, respectively.Through novel recycling method, both denitrification efficiency and SO2 oxidation ratio of the recycled catalyst are well recovered.The characterization results suggest that the highly polymerized vanadium species on the surface of the waste catalyst could not be removed by using conventional recycling method, but the novel recycling method is effective for removing these vanadium species and replacing them with highly dispersed vanadium species, resulting in a reduction of SO2 oxidation ratio of the recycled catalyst.
-
Key words:
- catalyst
- / waste
- / recycling
- / SO2 oxidation
-
选择性催化还原(SCR)技术是目前最成熟的烟气脱硝技术, 广泛用于燃煤电站等固定源的氮氧化物(NOx)排放控制, 其核心是SCR催化剂[1]。目前, 商用SCR催化剂一般是以V2O5为活性组分、WO3(或MoO3)为催化助剂、TiO2为载体的V2O5-WO3(MoO3)/TiO2催化剂。在实际使用过程中, 催化剂因活性位点被覆盖、流失或发生不可逆的化学转化而逐渐失活[2], 导致催化效率明显下降, 同时SO2氧化率显著上升。当催化剂不能满足SCR系统整体脱硝性能要求时, 即需进行更换, 被换下的催化剂根据失活和破损程度, 进行再生或者废弃处理, 而催化剂不能无限制再生, 最终都要进行废弃处理。
针对废弃SCR催化剂, 有学者提出采用物化方法集中处理, 回收钒、钨和钛等元素, 然而, 回收工艺涉及多种元素的分离提纯, 技术路线长且复杂, 还会引发诸多环境问题[3-5]。也有学者借鉴催化剂再生技术的思路, 通过对废弃催化剂进行有毒物质的清理以及活性组分的负载等工艺, 实现废弃催化剂的循环再利用。Lee等[6]采用0.5 mol/L的H2SO4溶液对废弃催化剂进行清洗, 随后采用0.05 mol/L的偏钒酸铵溶液和0.05 mol/L的偏钨酸铵溶液的混合液进行活性组分负载, 使催化剂的脱硝效率由处理前的45%提升至95%, 处理后催化剂表面绝大部分的有毒物质得以清除, 同时, SO42-的引入还增加了催化剂表面酸位点的数量和强度。史伟伟等[7]先用去离子水对废弃催化剂进行清洗, 随后进行活性组分浸渍, 发现当钒的再负载量达0.5%(质量分数)时, 可使脱硝效率由69.71%恢复至98.59%, 但SO2氧化率高达0.96%;为此尝试向催化剂中掺杂金属元素以控制SO2氧化率, 结果表明, 金属元素的掺杂能在一定程度内降低SO2氧化率, 但脱硝效率也随之降低。刘建华等[8]采用0.005 mol/L的H2SO4溶液对废弃催化剂进行处理, 随后进行活性组分负载, 结果表明, 催化活性有所恢复, 但SO2氧化率却由处理前的1.08%升高至1.18%。针对废弃催化剂的循环利用, 现有学者多将研究重点集中于催化活性的恢复上, 而忽略了SO2氧化率的控制问题, 而且仅有的少数研究也都表明处理后所得催化剂的SO2氧化率明显偏高。SO2氧化率是SCR催化剂的一项重要指标, 直接决定了SO3与硫酸铵盐的生成, 从而直接影响了催化剂硫中毒、空预器堵塞和设备腐蚀等问题[9]。
基于此, 本研究提出了一种新型的废弃催化剂再利用方法, 包括酸洗、还原酸浸以及活性组分负载等主要步骤, 在恢复催化剂脱硝效率的同时, 有效控制SO2氧化率。重点测试了再利用过程中不同的处理步骤对催化剂脱硝效率和SO2氧化率的影响, 并采用X射线荧光光谱分析(XRF)、物理吸附、X射线衍射(XRD)、氨气程序升温脱附(NH3-TPD)、氢气程序升温还原(H2-TPR)、X射线光电子能谱(XPS)和傅里叶变换红外光谱(FT-IR)等分析手段对催化剂进行表征。
1 实验部分
1.1 废弃及新鲜催化剂试样
实验所采用的废弃蜂窝V2O5-WO3/TiO2催化剂取自中国某电厂, 照片见图 1。实验前先进行分拣, 选择结构较为完整的催化剂模块, 切割出高50 mm的3×3孔催化剂块体备用。
图1
废弃SCR脱硝催化剂
Figure1.
Waste SCR denitrification catalyst
实验中新鲜催化剂采用等体积浸渍法制备, 具体方法如下:将适量的偏钒酸铵和偏钨酸铵分别配制成相应的盐溶液, 配制时向偏钒酸铵溶液中滴加适量的单乙醇胺以加速其溶解。然后将上述盐溶液依次加入到锐钛矿型纳米TiO2中, 搅拌均匀后超声震荡3 h, 并在室温下陈化24 h, 然后依次在110 ℃下干燥8 h, 550 ℃下焙烧3 h后获得新鲜催化剂。
1.2 废弃催化剂再利用技术
1.2.1 废弃催化剂常规再利用技术
针对上述废弃催化剂(记为1#), 按照如下方法进行常规再利用处理。首先进行物理除灰, 随后用去离子水在45 ℃下超声震荡清洗30 min, 然后用足量的0.5 mol/L的H2SO4溶液超声震荡清洗15 min, 最后在110 ℃下干燥8 h, 干燥后研磨筛分为100-200目的催化剂粉末(记为2#)。针对2#催化剂, 采用等体积浸渍法进行活性组分负载, 将适量的偏钒酸铵和偏钨酸铵分别配制成相应的盐溶液, 然后依次加入到2#催化剂粉末中, 搅拌均匀后超声震荡3 h, 并在室温下陈化24 h, 然后依次在110 ℃下干燥8 h, 400 ℃下焙烧3 h, 获得常规再利用催化剂(记为3#)。
1.2.2 废弃催化剂新型再利用技术
在上述常规处理技术的基础上, 本研究提出在酸洗之后采用还原酸浸法对废弃催化剂进行处理, 随后再进行活性组分再负载, 具体操作步骤如下。在搅拌条件下, 将适量质量浓度为98%的乙二醇和前述2#催化剂粉末(乙二醇与2#催化剂中活性组分五氧化二钒的物质的量比在5 :1-10 :1)加入到2 mol/L的H2SO4溶液中, 在95 ℃下还原酸浸处理4 h, 随后进行抽滤并用去离子水将滤渣漂洗至pH值为7, 最后在110 ℃下干燥8 h, 干燥后研磨筛分为100-200目的催化剂粉末(记为4#)。对4#催化剂进行活性组分负载, 方法同上, 由此获得再利用催化剂(记为5#)。此外, 将新鲜催化剂记为6#, 该新鲜催化剂中的V2O5和WO3的质量分数与3#和5#催化剂相当。
1.3 催化剂性能评价
催化剂的性能评价主要包括脱硝效率和SO2氧化率测试, 两者均在常压连续流动固定床石英管(内径为8 mm)反应器中进行。实验装置示意图见图 2, 主要由钢瓶气源、质量流量计、混气罐、SCR脱硝反应器、尾气处理系统、移动式全红外烟气分析仪、温控系统和给水系统组成。此外, 在测试SO2氧化率时还需在反应器入口和出口加装相应的SO3吸收装置。
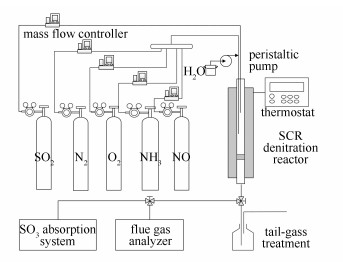 图2
催化剂性能评价系统示意图
Figure2.
Schematic of evaluation system for denitrification catalyst
图2
催化剂性能评价系统示意图
Figure2.
Schematic of evaluation system for denitrification catalyst
实验时, 催化剂置于脱硝反应器中, 反应所需的模拟烟气由NO、NH3、SO2、O2、水蒸气和N2组成, 各气体的流量由质量流量计精确调节, 水的流量经蠕动泵控制。反应气NO、NH3、O2和N2在混气罐中混合后与SO2分别流向反应器, 以避免NH3与SO2的直接反应。利用移动式全红外烟气分析仪(德国MRU公司, 型号MGA5)在线监测反应器进出口各气体浓度的变化。采用体积分数80%的异丙醇-水溶液对烟气中的SO3气体进行吸收, 随后以钍试剂为指示剂, 采用高氯酸钡标准溶液滴定吸收液中SO42-的浓度, 并根据烟气流量计算SO3的浓度。
典型烟气工况:温度为380 ℃, NO为402 mg/m3, NH3为228 mg/m3, SO2为858 mg/m3, O2为3%(体积分数), H2O为5%(体积分数), 平衡气为N2, 空速为54000 h-1, 总流量为2.70 L/min。实验中, 催化剂的脱硝效率η和SO2氧化率x分别用式(1)和式(2)进行计算。
式中, φNOin和φNOout分别为催化反应前后烟气中的NO浓度, 单位为mg/m3; φSO3, out为催化反应后烟气中的SO3浓度, 单位为mg/m3; φSO2, in为催化反应前烟气中的SO2浓度, 单位为mg/m3。
1.4 催化剂的表征
采用XRF-1800型X射线荧光光谱仪对样品进行XRF分析, 确定催化剂表面元素组成及相对含量, X射线管靶为铑靶(Rh), 管压为60 kV(Max), 检测浓度为10-6-100%, 最小分析微区直径为250 μ m, 最大扫描速率为300(°)/min。
采用ASAP2020型物理吸附仪对样品进行N2吸附-脱附实验, 测试催化剂的比表面积和孔结构, 测试前先将样品置于90 ℃下真空处理1 h, 紧接着在200 ℃下真空处理6 h; 然后在-196 ℃下采用静态法测量样品的吸附-脱附等温线, 由BET方程和BJH方程分别计算样品的比表面积和孔结构参数。
采用Bruker D8 ADVANCE型X射线粉末衍射仪对样品进行XRD分析, 确定催化剂表面物质的组成及物相, 采用Cu Kα辐射(λ = 0.15406 nm), 10°-90°扫描, 扫描速率为6(°)/min, 扫描步长为0.02°。
采用ChemBET Pulsar TPR/TPD型多功能吸附分析仪对样品进行H2-TPR和NH3-TPD分析, 确定催化剂的氧化还原性能及表面酸性、活性成分的分散度等性能, 采用热导检测器(TCD)进行测试, 测试温度为80-700 ℃, 升温速率为10 ℃/min。
采用Thermo escalab 250Xi型能谱仪对样品进行XPS分析, 确定催化剂表面元素的价态和相对含量, 以单色Al Kα (hv = 1486.6 eV)为射线源, 分析结果用C 1s的结合能(Eb = 284.8 eV)进行校准。
采用Nicolet iS10型傅里叶红外光谱仪对催化剂表面吸附物种、反应中间物种及官能团进行分析, 光谱为4000-800 cm-1, 采样分辨率为4 cm-1, 扫描32次, 扫描速率约为1次/s, 采用1 mg样品与100 mg KBr混合压片制样, 并保证自支撑片表面光滑且完好无裂缝。
2 结果与讨论
2.1 催化剂的组成及其脱硝效率和SO2氧化率
2.1.1 不同催化剂的组成成分、脱硝效率和SO2氧化率
采用前述工艺对废弃SCR催化剂进行处理, 经不同步骤处理后所得催化剂的XRF分析结果见表 1, 脱硝效率和SO2氧化率测试结果见图 3。商业蜂窝SCR脱硝催化剂中V2O5和WO3的质量分数一般约为1%和5%, 基于此, 自行制备了新鲜催化剂(6#)作为对照。其中, V2O5和WO3的质量分数分别为1.09%和5.08%。由表 1可知, 与新鲜催化剂(6#)相比, 废弃催化剂(1#)中V2O5的含量显著降低至0.56%, 而WO3的含量略微降低至4.61%, 可能是由于催化剂运行过程中活性组分化学转化为可挥发或可升华的络合物而流失[10], 是导致催化剂失活的主要原因之一。另外, 1#催化剂中还检测到相当量的碱金属、碱土金属和氧化铁等杂质, 这也是催化剂中毒失活的主要原因之一。因此, 1#催化剂的脱硝效率显著低于6#催化剂(77.0%和99.0%)。虽然, 1#催化剂中V2O5含量显著降低, 但其SO2氧化率却高于6#催化剂(0.46%和0.43%), 推测可能是由于催化剂长期遭受热力冲击, 导致表面钒物种迁移团聚, 形成高聚态的钒物种, 而这类高聚态钒物种又是催化SO2氧化的主要活性成分[11], 这一推论将在后续的NH3-TPD表征结果中进行论证。
 表 1
催化剂中各组分的含量
Table 1.
Content of each component in the catalysts
表 1
催化剂中各组分的含量
Table 1.
Content of each component in the catalysts
Catalyst Content w/% TiO2 V2O5 WO3 SiO2 Al2O3 BaO SO3 K2O Na2O Fe2O3 CaO 1# 80.75 0.56 4.61 3.95 3.13 2.51 1.71 0.18 0.08 0.76 1.55 2# 84.54 0.52 4.60 3.75 2.97 2.42 0.86 0.02 - 0.04 0.16 3# 83.58 1.07 5.06 3.75 2.95 2.41 0.85 0.02 - 0.04 0.15 4# 85.63 0.12 4.56 3.68 2.88 2.23 0.86 - - - - 5# 84.15 1.10 5.12 3.66 2.86 2.22 0.85 - - - - 6# 93.77 1.09 5.08 - - - 0.06 - - - - 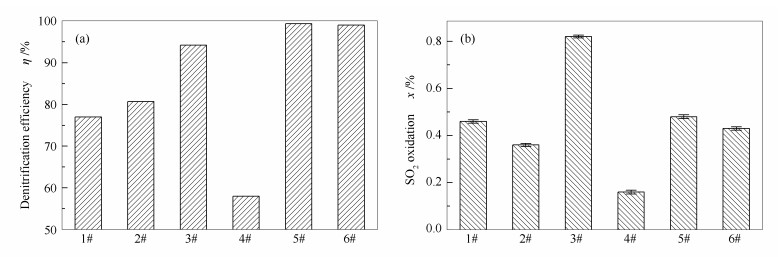 图3
不同催化剂脱硝效率和SO2氧化率
Figure3.
Denitrification efficiency(a) and SO2 oxidation ratio(b) of different catalysts
图3
不同催化剂脱硝效率和SO2氧化率
Figure3.
Denitrification efficiency(a) and SO2 oxidation ratio(b) of different catalysts
经酸洗后所得催化剂(2#)中V2O5和WO3的含量略有降低, 分别为0.52%和4.60%, 且杂质含量显著下降, 说明酸洗会使V2O5和WO3发生部分溶解而损失, 同时有效清除催化剂中的杂质。基于此, 2#催化剂的脱硝效率和SO2氧化率分别为80.7%和0.36%。在2#催化剂上负载活性组分, 所获得的3#催化剂中V2O5和WO3的含量分别恢复为1.07%和5.06%, 与6#催化剂基本相当; 3#催化剂的脱硝效率为94.2%, 活性基本得以恢复; 但其SO2氧化率却显著提升至0.80%, 明显高于6#催化剂的0.43%, 这是由于酸洗不能将催化剂中的高聚态钒物种清除, 而活性组分负载又进一步加剧了钒物种的团聚行为, 使得催化剂表面高聚态钒物种的数量显著增加。
采用新型再利用技术对2#催化剂进行还原酸浸处理, 由于还原酸浸可以有效溶解V2O5, 所得4#催化剂中V2O5的含量由0.52%显著降低至0.12%;而WO3的含量略微降低至4.56%, 同时催化剂表面残留的碱金属、碱土金属、氧化铁等有毒物质全部得以清除。由此可见, 还原酸浸处理能有效清除催化剂表面绝大部分的钒物种和有毒物质, 该催化剂脱硝效率和SO2氧化率均显著降低, 分别为58.0%和0.16%。继续对4#催化剂进行活性组分负载, 所得5#催化剂中V2O5和WO3的含量分别为1.10%和5.12%, 与新鲜催化剂相当; 该催化剂的脱硝效率为99.3%, 活性完全得以恢复; 更为重要的是, 其SO2氧化率仅为0.48%, 明显低于常规处理所得3#催化剂的0.80%。可见采用本研究提出的新型再利用技术对废弃催化剂进行处理不仅能恢复其脱硝效率, 还能有效控制SO2氧化率。
2.1.2 反应温度对再利用催化剂性能的影响
由上述结果可知, 采用新型再利用技术处理废弃催化剂, 在恢复脱硝效率的同时, 还可有效控制SO2氧化率, 是一种高效实用的废弃催化剂再利用技术。进一步考察了再利用催化剂在更高空速下对反应温度变化的适应性, 结果见图 4。
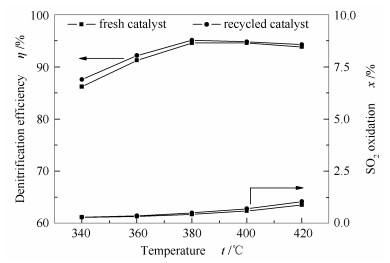 图4
温度对新鲜和再利用催化剂脱硝效率和SO2氧化率的影响
Figure4.
Effect of temperature on the denitrification efficiency and SO2 oxidation ratio of fresh and recycling catalysts(space velocity 162000 h-1, NH3/NO= 1.0)
图4
温度对新鲜和再利用催化剂脱硝效率和SO2氧化率的影响
Figure4.
Effect of temperature on the denitrification efficiency and SO2 oxidation ratio of fresh and recycling catalysts(space velocity 162000 h-1, NH3/NO= 1.0)
随着温度的升高, 再利用催化剂的脱硝效率呈现先增后减的趋势, 但在较宽的温度范围内均保持较高的脱硝效率。而SO2氧化率随温度的升高逐渐增大, 且温度越高增速越快, 这说明SO2氧化反应为典型的温度敏感性反应[12]。此外, 由图 4还可以看出, 经再利用处理后催化剂的脱硝效率不仅得以恢复, 而且在各温度条件下均略微优于新鲜催化剂, 这可能是由于再利用过程中使用的硫酸溶液引入了部分酸性位点, 参与了NH3的吸附与活化过程, 这一点将在后续的FT-IR表征结果中进一步说明。整体而言, 新型再利用催化剂的脱硝效率和SO2氧化率随温度的变化趋势与新鲜催化剂基本相同, 说明其对温度变化具有良好的适应性。
2.2 催化剂的表征
基于以上实验研究可知, 采用本研究所述方法获得的再利用催化剂具有优异的脱硝性能和较低的SO2氧化率, 为进一步探究再利用催化剂的理化性质及其与催化剂脱硝效率和SO2氧化率之间的关系, 对典型催化剂进行了分析表征。
2.2.1 物理结构分析
表 2为不同催化剂的比表面积、孔容和平均孔径的测定结果。
表 2
催化剂的比表面积和孔结构
Table 2.
Surface areas and pore structures of the catalysts
Catalyst Surface area A/(m2·g-1) Pore volume v/(cm3·g-1) Average pore diameter d/nm 1# 43.7 0.24 22.0 2# 53.4 0.26 19.8 3# 53.4 0.17 12.4 4# 61.2 0.21 13.6 5# 55.8 0.20 13.6 6# 79.4 0.37 18.6 由表 2可知, 与新鲜催化剂(6#)相比, 废弃催化剂(1#)的比表面积和孔容均显著降低, 平均孔径增大, 这是由于运行过程中催化剂长期经受飞灰沉积和热力冲击, 造成孔道堵塞及结构坍塌[13]。经酸洗后2#催化剂的孔结构得到一定程度的改善, 由前述XRF表征结果(表 1)可知, 这是由于表面沉积的碱金属、氧化铁和硫酸盐等杂质被清除的缘故。与2#催化剂相比, 经活性组分负载的3#催化剂的比表面积略微降低, 但由于2#催化剂的孔径较大, 因而在负载过程中大量的活性成分聚集在这些直径较大的孔内难以分散, 使得孔容降低的同时平均孔径显著降低, 这也可能是常规再利用3#催化剂SO2氧化率偏高的原因之一。对2#催化剂进行还原酸浸处理, 所得4#催化剂与处理前相比孔容略有降低, 但由于所采用的酸浸液为高浓度的H2SO4溶液, 对催化剂表面具有极强的刻蚀作用, 从而形成大量直径较小的孔结构, 导致平均孔径减小而比表面积增大[14]。随后对4#催化剂进行活性组分负载, 所得5#催化剂的比表面积有所降低, 而孔容和平均孔径基本没有变化, 这是由于还原酸浸处理后4#催化剂的孔径趋于均匀且相对较小, 避免了活性成分的聚集, 使之在催化剂表面高度分散, 催化剂活性得以恢复, 同时SO2氧化率得以控制。
2.2.2 表面物相分析
图 5为不同催化剂的XRD谱图。所有催化剂样品均有明显的锐钛矿型TiO2衍射峰(PDF-ICDD 65-5714), 同时除新鲜催化剂(6#)外, 其余催化剂(1#-5#)还有BaSO4的衍射峰(PDF-ICDD 24-1035), 这是由于本研究所采用的废弃催化剂在成型过程中, 添加了BaSO4的缘故。值得注意的是, 所有样品中均未显示钒和钨的衍射峰, 这可能是由于钒和钨在TiO2表面高度分散或含量过低[15]。
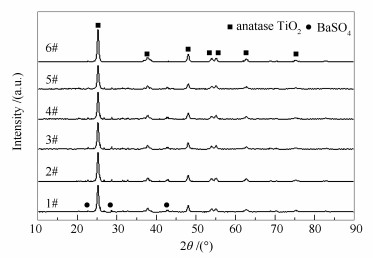 图5
催化剂的XRD谱图
Figure5.
XRD patterns of the catalysts
图5
催化剂的XRD谱图
Figure5.
XRD patterns of the catalysts
2.2.3 表面酸性及钒物种分散度分析
图 6为不同催化剂的NH3-TPD谱图。所有催化剂在200-350 ℃均出现一个较宽的脱附峰, 在350-450 ℃出现一个微弱的脱附峰, 分别归属于催化剂表面的Brønsted酸性位和Lewis酸性位[16], 可见在SCR脱硝反应中Brønsted酸位是主要的酸性中心。与新鲜催化剂(6#)相比, 废弃催化剂(1#)的低温脱附峰向低温方向偏移且峰面积显著减小, 说明表面酸性位的强度和数量均降低, 可能是由于碱金属、碱土金属等物质的沉积使得表面酸性被中和, 以及运行过程中活性组分由于生成可挥发或可升华的络合物而流失。经酸洗后2#催化剂的表面酸性位在强度和数量上均有所恢复, 结合XRF表征结果可知, 是由于上述杂质被有效清除的缘故。直接进行活性组分负载后的3#催化剂表面酸性位的强度和数量均有所提升, 因而催化活性基本得以恢复。若对2#催化剂进行还原酸浸处理, 所得4#催化剂的脱附峰的出峰位置和峰面积均显著下降, 结合XRF表征结果可知, 是由于表面钒物种被清除的缘故。对4#催化剂进行活性组分负载, 所得再利用催化剂(5#)的脱附峰的出峰位置和峰面积均得到显著提升, 这说明催化剂表面酸性位的强度和数量均得以恢复。
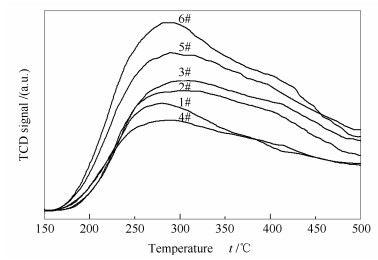 图6
催化剂的NH3-TPD谱图
Figure6.
NH3-TPD profiles of the catalysts
图6
催化剂的NH3-TPD谱图
Figure6.
NH3-TPD profiles of the catalysts
此外, 对比催化剂NH3脱附峰的峰面积可定性比较V2O5在催化剂表面的分散度, 进而分析其存在形态, 以了解其在SCR反应和SO2氧化反应中的性能。研究表明[17, 18], 催化剂表面Brønsted酸性位的数量与其上钒物种的分散度呈正比关系, 即催化剂表面钒物种的分散度越低, 暴露出的V5+-OH数量越少, NH3的吸附量也越少, 在NH3-TPD谱图中则表现为脱附峰的峰面积越小。由图 6可知, 常规再利用催化剂(3#)与新型再利用催化剂(5#)中活性组分V2O5的含量相当, 但前者的脱附峰面积显著低于后者, 说明3#催化剂表面钒物种的分散度低于5#催化剂, 即3#催化剂表面钒物种团聚现象严重, 必然导致高聚态钒物种的形成, 由此证实前述推论, 即催化剂表面高聚态钒物种的存在导致催化剂SO2氧化率偏高。而5#催化剂表面的钒物种呈高度分散状态, 且这种高度分散的单体或低聚态的钒氧物种并非SO2氧化的主要活性物种, 因此, 其SO2氧化率得以控制。
2.2.4 氧化还原性能分析
图 7为不同催化剂的H2-TPR谱图。由图 7可知, 所有催化剂均在400-600 ℃呈现一个明显的还原峰, 对应于催化剂表面V5+到V4+的还原[19]。新鲜催化剂(6#)的还原峰中心温度为475 ℃, 废弃催化剂(1#)的还原峰向高温方向迁移至542 ℃, 说明表面钒物种的还原性发生了突降, 可能是由于催化剂表面碱金属和碱土金属等杂质与钒结合, 导致还原性降低[20]。经酸洗后2#催化剂的还原性在很大程度上得以恢复, 同时其还原峰面积显著降低, 说明酸洗过程可能会破坏表面原子间的化学键, 导致相互间作用力减弱。直接负载活性组分的3#催化剂, 还原性略有提升。对2#催化剂进行还原酸浸处理所得4#催化剂的还原峰中心温度提高, 还原性有所下降, 这在该工艺中为正面效应, 说明了还原酸浸过程确实能清除催化剂表面残留的高聚态钒物种。对4#催化剂进行活性组分负载所得再利用催化剂(5#)的还原峰向低温方向迁移至482 ℃, 与废弃催化剂(1#)相比显著降低, 说明经再利用处理后表面沉积的杂质得以清除, 同时表面钒物种的形态得以优化, 使得氧化还原性能适中, 一方面, 使催化剂剂活性得以恢复; 另一方面, 使SO2氧化率得以控制。
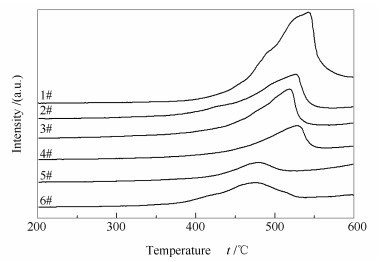 图7
催化剂的H2-TPR谱图
Figure7.
H2-TPR profiles of the catalysts
图7
催化剂的H2-TPR谱图
Figure7.
H2-TPR profiles of the catalysts
2.2.5 表面元素价态及相对含量分析
图 8为不同催化剂的V 2p3/2 XPS谱图, 催化剂表面V5+和V4+的相对含量见表 3。由图 8可知, 结合能为515.7-516.3 eV的谱峰归属于V4+, 结合能为516.9-517.3 eV的谱峰归属于V5+[21]。由表 3可知, 新鲜催化剂(6#)表面V5+的相对含量为66.00%, 废弃催化剂(1#)表面V5+的相对含量显著降低为13.31%, 这应该是由于在运行过程中活性组分V2O5发生了还原反应, 例如与硫物种反应生成VOSO4等不具有SCR催化活性的物质[22], 因而废弃催化剂(1#)活性显著降低, 这也说明了催化剂的活性与表面V5+含量有着紧密的联系, 在一定范围内两者呈正相关[23]。经酸洗后2#催化剂中V5+的相对含量回升至22.51%。直接进行活性组分负载后的3#催化剂表面V5+的相对含量进一步提升至57.28%, 这与其催化活性恢复的趋势相一致。对2#催化剂进行还原酸浸处理所得的4#催化剂的表面检测不到钒物种, 再次说明采用还原酸浸法可有效清除催化剂表面的钒物种。对4#催化剂进行活性组分负载, 所得再利用催化剂(5#)表面V5+的相对含量达64.84%, 这一适中的V5+相对含量有利于催化过程中氧化还原循环反应的进行。
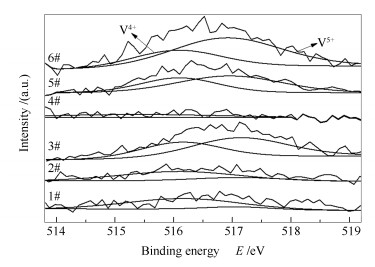 图8
催化剂的V 2p3/2 XPS谱图
Figure8.
XPS spectra of V 2p3/2 for the catalysts
表 3
催化剂中V5+、V4+、Oα和Oβ的相对含量
Table 3.
Relative contents of V5+, V4+, Oα and Oβ in the catalysts
图8
催化剂的V 2p3/2 XPS谱图
Figure8.
XPS spectra of V 2p3/2 for the catalysts
表 3
催化剂中V5+、V4+、Oα和Oβ的相对含量
Table 3.
Relative contents of V5+, V4+, Oα and Oβ in the catalysts
Catalyst Relative content/% V5+/(V5++V4+) V4+/(V5++V4+) Oα/(Oα+Oβ) Oβ/(Oα+Oβ) 1# 13.31 86.69 15.05 84.95 2# 22.51 77.49 25.34 74.66 3# 57.28 42.72 40.25 59.75 4# - - 28.77 71.23 5# 64.84 35.16 42.54 57.46 6# 66.00 34.00 47.38 52.62 图 9为不同催化剂的O 1s XPS谱图, 催化剂表面Oα和Oβ的相对含量见表 3。由图 9可知, 结合能位于529.5-531 eV的谱峰归属于晶格氧(O2-, 记为Oβ), 位于531-532.5 eV的谱峰归属于化学吸附氧(O2-, 记为Oα)[24], 化学吸附氧比晶格氧更具有活性, 在SCR反应中更为重要。新鲜催化剂(6#)表面Oα的相对含量为47.38%, 废弃催化剂(1#)表面Oα的相对含量降低为15.05%, 这可能是由于杂质的沉积占据了催化剂表面氧空位, 并在氧空位形成强吸附的缘故[25]。此外, 催化剂表面钒物种磨损流失和高温烧结也会导致表面氧空位的数量急剧下降。经酸洗后2#催化剂表面Oα的相对含量回升至25.34%。直接进行活性组分负载后的3#催化剂表面Oα的相对含量进一步提升至40.25%, 这与其催化活性基本得以恢复相一致。对2#催化剂进行还原酸浸处理, 所得4#催化剂表面Oα的相对含量显著降低, 这与催化剂表面钒物种被清除, 导致表面氧空位数量减少有关。对4#催化剂进行活性组分负载, 所得再利用催化剂(5#)表面Oα的相对含量回升至42.54%, 这是因为再负载的钒物种高度分散, 在催化剂的表面造成电子失衡、空缺和不饱和现象, 进而增加化学吸附氧削弱晶格氧, 使其表面化学吸附氧和晶格氧的相对含量适中, 有利于催化反应过程中各种氧物种间的转化。
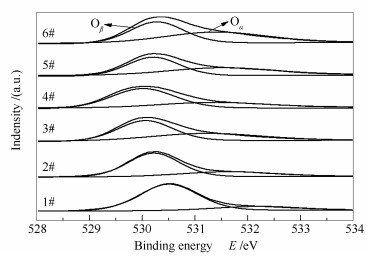 图9
催化剂的O 1s XPS谱图
Figure9.
XPS spectra of O 1s for the catalysts
图9
催化剂的O 1s XPS谱图
Figure9.
XPS spectra of O 1s for the catalysts
2.2.6 表面物种及官能团分析
图 10为不同催化剂的FT-IR谱图。由图 10可知, 所有催化剂在3418和1634 cm-1处均出现了两个较宽的特征峰, 它们分别归属于O-H的拉伸振动和H-O-H的弯曲振动[26], 说明催化剂表面吸附有水分子, 且与样品表面的羟基之间形成了大量氢键, 其原因可能是样品干燥不彻底。研究表明[27], 过多的表面羟基对催化活性可能会有不利影响, 但适量的羟基在催化剂表面可能活化活性位点。除4#催化剂外, 其余催化剂在1376 cm-1处均出现一个微弱的归属于Brønsted酸性位(V-OH基团)的特征峰[28], 这说明经还原酸浸处理后, 废弃催化剂表面残留的钒物种已被清除; 且再利用催化剂(5#)和新鲜催化剂(6#)在该位置的特征峰相对比较明显, 说明经再利用处理后催化剂表面的钒物种得以恢复。由图 10还可看出, 除新鲜催化剂(6#)外, 其余样品的谱图中在1080、1124和1184 cm-1出现三个特征峰, 这归结于催化剂表面SO42-由Td对称性构型向C2v对称性构型的转变[29]。值得注意的是, 虽然废弃催化剂(1#)与2#-5#催化剂的谱图中均出现了SO42-的特征峰, 但是它们的来源不同, 因而对催化剂的影响也不同:前者来源于催化剂运行过程中硫酸盐物种的沉积, 从而堵塞了表面活性位点, 引发催化剂中毒; 而后者来源于再利用过程中使用的H2SO4溶液, 由此引入的SO42-在可作为弱酸性位点参与NH3的吸附与活化过程[30], 这与再利用催化剂(5#)的脱硝效率略高于新鲜催化剂(6#)这一结果相吻合。与其余催化剂不同的是, 新鲜催化剂(6#)在1048和1134 cm-1处还出现了明显的归属于Lewis酸性位(V=O基团)的特征峰[31], 这与NH3-TPD谱图中在350-450 ℃出现的较为明显的对应于Lewis酸性位的脱附峰相一致。
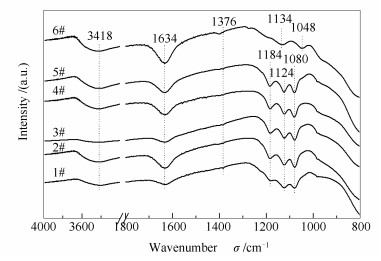 图10
催化剂的FT-IR谱图
Figure10.
FT-IR spectra of the catalysts
图10
催化剂的FT-IR谱图
Figure10.
FT-IR spectra of the catalysts
3 结论
基于废弃SCR脱硝催化剂循环再利用的技术需求, 提出了一种新型的再利用处理工艺, 包括酸洗、还原酸浸和活性组分负载等步骤。研究结果表明, 废弃催化剂(1#)中V2O5的含量显著低于新鲜催化剂(6#)(0.56%和1.09%), WO3的含量也有所降低(4.61%和5.08%), 同时还含有相当量的碱金属、碱土金属和氧化铁等杂质, 一方面, 使得催化剂表面Brønsted酸性位的强度、数量以及钒物种的氧化还原性能降低, 同时还使得表面V5+和Oα的相对含量下降, 因而废弃催化剂(1#)的脱硝效率显著低于新鲜催化剂(6#)(77.0%和99.0%)。此外, 催化剂表面钒物种在运行过程中长期遭受热力冲击而迁移团聚, 形成高聚态的钒物种, 导致其SO2氧化率反而高于新鲜催化剂(6#)(0.46%和0.43%)。
废弃催化剂(1#)经酸洗后所得2#催化剂中的杂质基本得以清除, 而V2O5和WO3的质量分数因发生部分溶解而分别降低为0.52%和4.60%。对2#催化剂直接进行活性组分负载所得常规再利用催化剂(3#)中V2O5和WO3的质量分数分别恢复为1.07%和5.06%, 脱硝效率恢复为94.2%, 但由于仅进行酸洗不能将催化剂中高聚态的钒物种清除, 同时直接进行活性组分负载会加剧钒物种的团聚作用, 其SO2氧化率高达0.80%, 明显高于新鲜催化剂(6#)的0.43%, 可见常规再利用技术不能有效控制其SO2氧化率。
对2#催化剂进行还原酸浸处理, 所得4#催化剂中V2O5大量溶解, 其含量由0.52%显著降低至0.12%, 而WO3的含量略微降低至4.56%, 同时催化剂表面残留的少量杂质全部得以清除。继续对4#催化剂进行活性组分负载, 所得新型再利用催化剂(5#)中V2O5和WO3的质量分数恢复为1.10%和5.12%, 同时催化剂表面Brønsted酸性位的强度、数量以及钒物种的氧化还原性能基本得以恢复, V5+和Oα的相对含量显著提升, 因而其脱硝效率恢复为99.3%, 且SO2氧化率仅为0.48%, 明显低于常规再利用催化剂(3#)的0.80%。
-
-
[1]
黄增斌, 李翠清, 王振, 徐胜美, 冯凌波, 王虹, 宋永吉, 张伟. 不同分子筛负载锰铈催化剂的低温NH3-SCR脱硝性能[J]. 燃料化学学报, 2016,44,(11): 495-499. HUANG Zeng-bin, LI Cui-qing, WANG Zhen, XU Sheng-mei, FENG Ling-bo, WANG Hong, SONG Yong-ji, ZHANG Wei. Performance of Mn-Ce catalysts supported on different zeolites in the NH3-SCR of NOx[J]. J Fuel Chem Technol, 2016, 44(11): 495-499.
-
[2]
商雪松, 陈进生, 赵金平, 张福旺, 徐亚, 徐琪. SCR脱硝催化剂失活及其原因研究[J]. 燃料化学学报, 2011,39,(6): 465-470. SHANG Xue-song, CHEN Jin-sheng, ZHAO Jin-ping, ZHANG Fu-wang, XU Ya, XU Qi. Discussion on the deactivation of SCR denitrification catalyst and its reasons[J]. J Fuel Chem Technol, 2011, 39(6): 465-470.
-
[3]
HUO Y T, CHANG Z D, LI W J, LIU S X, DONG B. Reuse and valorization of vanadium and tungsten from waste V2O5-WO3/TiO2 SCR catalyst[J]. Waste Biomass Valor, 2015, 6(2): 159-165. doi: 10.1007/s12649-014-9335-2
-
[4]
ZHAO Z P, GUO M, ZHANG M. Extraction of molybdenum and vanadium from the spent diesel exhaust catalyst by ammonia leaching method[J]. J Hazard Mater, 2015, 286: 402-409. doi: 10.1016/j.jhazmat.2014.12.063
-
[5]
陈晨, 陆强, 蔺卓玮, 李文艳, 董长青. 燃煤电厂废弃SCR脱硝催化剂元素回收研究进展[J]. 化工进展, 2016,35,(10): 3306-3312. CHEN Chen, LU Qiang, LIN Zhuo-wei, LI Wen-yan, DONG Chang-qing. Research progress of element recovery of waste De-NOx SCR catalyst from coal-fired power plants[J]. Chem Ind Eng Prog, 2016, 35(10): 3306-3312.
-
[6]
LEE J B, LEE T W, SONG K C, LEE I Y. Method of regenerating honeycomb type SCR catalyst by air lift loop reactor: US, 2006/0094587A1[P]. 2006-05-04.
-
[7]
史伟伟. SCR脱硝催化剂再生浸渍及其SO2氧化控制[D]. 广州: 华南理工大学, 2013.SHI Wei-wei. Impregnation regeneration and SO2 oxidation control of the SCR denitrification catalysts[D]. Guangzhou: South China University of Technology, 2013.
-
[8]
刘建华. 平板式SCR脱硝催化剂再生及其SO2氧化控制[D]. 广州: 华南理工大学, 2016.LIU Jian-hua. Regeneration and SO2 oxidation control of plate-type SCR denitrification catalysts[D]. Guangzhou: South China University of Technology, 2016.
-
[9]
MENASHA J, DUNN-RANKIN D, MUZIO L, STALLINGS J. Ammonium bisulfate formation temperature in a bench-scale single-channel air preheater[J]. Fuel, 2011, 90(7): 2445-2453. doi: 10.1016/j.fuel.2011.03.006
-
[10]
YAKHNIN V Z, MENZINGER M. Deactivation avalanches through the interaction of locally deactivated catalyst with traveling hot spots[J]. Stud Surf Sci Catal, 1999, 126: 291-298. doi: 10.1016/S0167-2991(99)80478-9
-
[11]
DUNN J P, STENGER H G, WACHS I E. Oxidation of SO2 over supported metal oxide catalysts[J]. J Catal, 1999, 181(2): 233-243. doi: 10.1006/jcat.1998.2305
-
[12]
ORSENIGO, LIETTI, TRONCONI, FORZATTI, BREGANI. Dynamic investigation of the role of the surface sulfates in NOx reduction and SO2 oxidation over DeNOx-SCR catalysts[J]. Ind Eng Chem Res, 1998, 165(37): 2350-2359.
-
[13]
LIU X S, WU X D, XU T F, WENG D, SI Z C, RAN R. Effects of silica additive on the NH3-SCR activity and thermal stability of a V2O5/WO3-TiO2 catalyst[J]. Chin J Catal, 2016, 37(8): 1340-1346. doi: 10.1016/S1872-2067(15)61109-3
-
[14]
徐浩, 延卫, 游莉. 不同酸处理对钛基体性能的影响[J]. 稀有金属材料与工程, 2011,40,(9): 1550-1554. XU Hao, YAN Wei, YOU Li. Effects of various acids treatment on the properties of titanium substrate[J]. Rare Met Mater Eng, 2011, 40(9): 1550-1554.
-
[15]
刘俊, 王亮亮, 费兆阳, 陈献, 汤吉海, 崔咪芬, 乔旭. 非晶态CeO2@TiO2催化剂的结构、性质及其选择催化还原脱硝性能[J]. 燃料化学学报, 2016,44,(8): 954-960. LIU Jun, WANG Liang-liang, FEI Zhao-yang, CHEN Xian, TANG Ji-hai, CUI Mi-fen, QIAO Xu. Structure and properties of amorphous CeO2@TiO2 catalyst and its performance in the selective catalytic reduction of NO with NH3[J]. J Fuel Chem Technol, 2016, 44(8): 954-960.
-
[16]
FANG C, ZHANG D S, CAI S X, ZHANG L, HUANG L, LI H R, MAITARAD P, SHI L Y, GAO R H, ZHANG J P. Low-temperature selective catalytic reduction of NO with NH3 over nanoflaky MnOx on carbon nanotubes in situ prepared via a chemical bath deposition route[J]. Nanoscale, 2013, 05(19): 9199-9207. doi: 10.1039/c3nr02631k
-
[17]
RAMIS G, BUSCA G, LORENZELLL V. Spectroscopic characterization of magnesium vanadate catalysts. Part 2. FTIR study of the surface properties of pure and mixed-phase powders[J]. J Chem Soc Faraday Trans, 1994, 90(9): 1293-1299. doi: 10.1039/FT9949001293
-
[18]
LIU X, LI J H, LI X, PENG Y, WANG H, JIANG X M, WANG L W. NH3 selective catalytic reduction of NO:A large surface TiO2 support and its promotion of V2O5 dispersion on the prepared catalyst[J]. Chin J Catal, 2016, 37(6): 878-887. doi: 10.1016/S1872-2067(15)61041-5
-
[19]
PENG Y, LI J H, SI W Z, LUO J M, WANG Y, JIE F, LI X, CRITTENDEN J, HAO J M. Deactivation and regeneration of a commercial SCR catalyst:Comparison with alkali metals and arsenic[J]. Appl Catal B:Environ, 2015, 168-169: 195-202. doi: 10.1016/j.apcatb.2014.12.005
-
[20]
LI Q C, CHEN S F, LIU Z Y, LIU Q Y. Combined effect of KCl and SO2 on the selective catalytic reduction of NO by NH3 over V2O5/TiO2 catalyst[J]. Appl Catal B:Environ, 2015, 164: 475-482. doi: 10.1016/j.apcatb.2014.09.036
-
[21]
DONG G J, ZHANG Y F, ZHAO Y, BAI Y. Effect of the pH value of precursor solution on the catalytic performance of V2O5-WO3/TiO2 in the low temperature NH3-SCR of NOx[J]. J Fuel Chem Technol, 2014, 42(12): 1455-1463. doi: 10.1016/S1872-5813(15)60003-2
-
[22]
杨剑, 林凡, 陈奎, 孔明, 赵冬, 孟飞. 含铈钒基SCR催化剂脱硝性能及SO2失活机理研究[J]. 燃料化学学报, 2016,44,(11): 1394-1400. doi: 10.3969/j.issn.0253-2409.2016.11.017YANG Jian, LIN Fan, CHEN Kui, KONG Ming, ZHAO Dong, MENG Fei. Activity and SO2 deactivation mechanism of vanadium series catalyst containing cerium[J]. J Fuel Chem Technol, 2016, 44(11): 1394-1400. doi: 10.3969/j.issn.0253-2409.2016.11.017
-
[23]
沈伯雄, 卢凤菊, 高兰君, 岳时吉. 中温商业SCR催化剂碱和碱土中毒特性研究[J]. 燃料化学学报, 2011,44,(4): 500-506. SHEN Bo-xiong, LU Feng-ju, GAO Lan-jun, YUE Shi-ji. Study on alkali and alkaline earths poisoning characteristics for a commercial SCR catalyst[J]. J Fuel Chem Technol, 2011, 44(4): 500-506.
-
[24]
LIU J, LI X Y, ZHAO Q D, KE J, XIAO H N, LV X J, LIU S M, TADÉ M, WANG S B. Mechanistic investigation of the enhanced NH3-SCR on cobalt-decorated Ce-Ti mixed oxide:In situ, FT-IR analysis for structure-activity correlation[J]. Appl Catal B:Environ, 2017, 200: 297-308. doi: 10.1016/j.apcatb.2016.07.020
-
[25]
沈伯雄, 熊丽仙, 刘亭, 王静, 田晓娟. 纳米负载型V2O5-WO3/TiO2催化剂碱中毒及再生研究[J]. 燃料化学学报, 2010,38,(1): 85-90. SHEN Bo-xiong, XIONG Li-xian, LIU Ting, WANG Jing, TIAN Xiao-juan. Alkali deactivation and regeneration of nano V2O5-WO3/TiO2 catalysts[J]. J Fuel Chem Technol, 2010, 38(1): 85-90.
-
[26]
ZHAO H, BENNICI S, SHEN J, AUROUX A. The influence of the preparation method on the structural, acidic and redox properties of V2O5-TiO2/SO42- catalysts[J]. Appl Catal A:Gen, 2009, 356(2): 121-128. doi: 10.1016/j.apcata.2008.12.037
-
[27]
EL-MAAZAWI M, FINKEN A N, NAIR A B, GRASSIAN V H. Adsorption and photocatalytic oxidation of acetone on TiO2:An in situ transmission FT-IR study[J]. J Catal, 2000, 191(1): 138-146. doi: 10.1006/jcat.1999.2794
-
[28]
姜烨, 高翔, 杜学森, 毛剑宏, 骆仲泱, 岑可法. PbO对V2O5/TiO2催化剂NH3选择性催化还原NO的影响[J]. 工程热物理学报, 2009,30,(11): 1973-1976. doi: 10.3321/j.issn:0253-231X.2009.11.048JIANG Ye, GAO Xiang, DU Xue-sen, MAO Jian-hong, LUO Zhong-yang, CEN Ke-fa. The effect of PbO on a V2O5/TiO2 catalyst for selective catalytic reduction on NO with NH3[J]. J Eng Thermophys, 2009, 30(11): 1973-1976. doi: 10.3321/j.issn:0253-231X.2009.11.048
-
[29]
CHEN J P, YANG R T. Selective catalytic reduction of NO with NH3 on SO42-/TiO2 superacid catalyst[J]. J Catal, 1993, 139(1): 277-288. doi: 10.1006/jcat.1993.1023
-
[30]
SHANG X S, HU G R, HE C, ZHAO J P, ZHANG F W, XU Y, ZHANG Y F, LI J R, CHEN J S. Regeneration of full-scale commercial honeycomb monolith catalyst (V2O5-WO3/TiO2) used in coal-fired power plant[J]. J Ind and Eng Chem, 2012, 18(1): 513-519. doi: 10.1016/j.jiec.2011.11.070
-
[31]
孔明, 刘清才, 赵东, 任山, 孟飞. NaCI与Hg0对V2O5-WO3/TiO2 SCR脱硝催化剂的协同作用研究[J]. 燃料化学学报, 2015,43,(12): 1504-1509. doi: 10.3969/j.issn.0253-2409.2015.12.015KONG Ming, LIU Qing-cai, ZHAO Dong, REN Shan, MENG Fei. Synergy of NaCl and Hg0 on V2O5-WO3/TiO2 SCR catalysts[J]. J Fuel Chem Technol, 2015, 43(12): 1504-1509. doi: 10.3969/j.issn.0253-2409.2015.12.015
-
[1]
-

图 3 不同催化剂脱硝效率和SO2氧化率
Figure 3 Denitrification efficiency(a) and SO2 oxidation ratio(b) of different catalysts

图 4 温度对新鲜和再利用催化剂脱硝效率和SO2氧化率的影响
Figure 4 Effect of temperature on the denitrification efficiency and SO2 oxidation ratio of fresh and recycling catalysts(space velocity 162000 h-1, NH3/NO= 1.0)
表 1 催化剂中各组分的含量
Table 1. Content of each component in the catalysts
Catalyst Content w/% TiO2 V2O5 WO3 SiO2 Al2O3 BaO SO3 K2O Na2O Fe2O3 CaO 1# 80.75 0.56 4.61 3.95 3.13 2.51 1.71 0.18 0.08 0.76 1.55 2# 84.54 0.52 4.60 3.75 2.97 2.42 0.86 0.02 - 0.04 0.16 3# 83.58 1.07 5.06 3.75 2.95 2.41 0.85 0.02 - 0.04 0.15 4# 85.63 0.12 4.56 3.68 2.88 2.23 0.86 - - - - 5# 84.15 1.10 5.12 3.66 2.86 2.22 0.85 - - - - 6# 93.77 1.09 5.08 - - - 0.06 - - - -  下载: 导出CSV
下载: 导出CSV
表 2 催化剂的比表面积和孔结构
Table 2. Surface areas and pore structures of the catalysts
Catalyst Surface area A/(m2·g-1) Pore volume v/(cm3·g-1) Average pore diameter d/nm 1# 43.7 0.24 22.0 2# 53.4 0.26 19.8 3# 53.4 0.17 12.4 4# 61.2 0.21 13.6 5# 55.8 0.20 13.6 6# 79.4 0.37 18.6
下载: 导出CSV
表 3 催化剂中V5+、V4+、Oα和Oβ的相对含量
Table 3. Relative contents of V5+, V4+, Oα and Oβ in the catalysts
Catalyst Relative content/% V5+/(V5++V4+) V4+/(V5++V4+) Oα/(Oα+Oβ) Oβ/(Oα+Oβ) 1# 13.31 86.69 15.05 84.95 2# 22.51 77.49 25.34 74.66 3# 57.28 42.72 40.25 59.75 4# - - 28.77 71.23 5# 64.84 35.16 42.54 57.46 6# 66.00 34.00 47.38 52.62
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 23
- 文章访问数: 6796
- HTML全文浏览量: 1171
 下载:
下载:
