引用本文:
舒庆, 唐国强, 刘峰生, 邹文强, 贺江凡. 新型Brönsted-Lewis酸性催化剂LaPW12O40/SiO2制备及其在催化酯化反应合成生物柴油中的应用[J]. 燃料化学学报,
2017, 45(8): 939-949.

Citation: SHU Qing, TANG Guo-qiang, LIU Feng-sheng, ZOU Wen-qiang, HE Jiang-fan. Preparation and application of a novel Brönsted-Lewis acid catalyst LaPW12O40/SiO2 for the synthesis of biodiesel via esterification reaction[J]. Journal of Fuel Chemistry and Technology, 2017, 45(8): 939-949.
Citation: SHU Qing, TANG Guo-qiang, LIU Feng-sheng, ZOU Wen-qiang, HE Jiang-fan. Preparation and application of a novel Brönsted-Lewis acid catalyst LaPW12O40/SiO2 for the synthesis of biodiesel via esterification reaction[J]. Journal of Fuel Chemistry and Technology, 2017, 45(8): 939-949.
新型Brönsted-Lewis酸性催化剂LaPW12O40/SiO2制备及其在催化酯化反应合成生物柴油中的应用
摘要:
以十二磷钨杂多酸(Tungstophosphoric acid,H3PW12O40)为基体,分别通过普通浸渍法、溶胶凝胶法和超声浸渍法进行了La3+改性作用,合成了三种固体酸催化剂A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40。采用X射线荧光光谱(XRF)、孔径比表面积测定、X射线粉末衍射(XRD)、透射电镜(TEM)、红外光谱(FT-IR)、热重(TG)、N2吸附-脱附、NH3程序升温脱附(NH3-TPD)、吡啶吸附红外光谱(Py-FTIR)、X射线光电子能谱(XPS)等方法对合成的催化剂进行了表征,并比较了以上催化剂在用于催化以油酸和甲醇为反应物经酯化反应合成生物柴油时的活性和稳定性。结果表明,B-LaPW12O40/SiO2具有最高催化活性,当甲醇与油酸的物质的量比为8:1,催化剂用量为反应物总质量的2%,反应温度为65 ℃,反应1 h后,油酸的转化率即高达93%。循环使用B-LaPW12O40/SiO2催化剂六次后,油酸的转化率仍高达86.4%。B-LaPW12O40/SiO2的高催化活性和稳定性可归因于在溶胶凝胶的转化过程中,作为硅源材料的四乙氧基硅(TEOS)易在酸性条件下发生水解反应形成SiO2网络,进而SiO2网络中的硅醇键与H3PW12O40中的H+发生配位作用,生成具有强静电吸附力的(≡Si-OH2+)(H2PW12O40-)络合物。随着该络合物的形成,促进了La3+在SiO2表面的吸附而堵塞了H3PW12O40的孔道结构,抑制了H3PW12O40颗粒在焙烧过程中进一步聚集长大。SiO2将作为载体并以干凝胶状态存在于B-LaPW12O40/SiO2催化剂中,由于SiO2凝胶的高比表面积而使B-LaPW12O40/SiO2具有了较大的比表面积,从H3PW12O40的1.4 m2/g增加至31.3 m2/g。并且,通过吡啶吸附红外光谱确定B-LaPW12O40/SiO2为Brönsted-Lewis酸型固体酸,由于Brönsted酸位易与酯化反应过程中生成的水发生水合反应而失活,因而Lewis酸位的形成有助于减少催化剂的失活现象发生。Lewis酸位的出现可归因于(≡Si-OH2+)(H2PW12O40-)与吸附在其表面的具有强吸电子作用的La3+发生键合作用后生成了LaPW12O40/SiO2。
-
关键词:
- 镧改性
- / 磷钨杂多酸
- / 溶胶-凝胶法
- / Brönsted-Lewis固体酸
- / 酯化反应
English
Preparation and application of a novel Brönsted-Lewis acid catalyst LaPW12O40/SiO2 for the synthesis of biodiesel via esterification reaction
Abstract:
In this study, H3PW12O40 (Tungstophosphoric acid) was applied as matrix, and which was modified by La3+ through conventional impregnation method, ultrasonic impregnation method and sol-gel method, obtained three solid acid catalysts: A-LaPW12O40, B-LaPW12O40/SiO2 and C-LaPW12O40. These above catalysts were characterized by X-ray fluorescence spectrometer, specific surface area and porosity analyzer, X-ray diffraction, transmission electron microscopy, Fourier transform infrared spectoscopy, thermogravimetric analysis, N2/adsorption-desorption, NH3 temperature programmed desorption, pyridine adsorption IR spectra and X-ray photoelectron spectroscopy. The catalytic activities and stabilities of them were compared when they were used for the catalytic synthesis of biodiesel from the esterification reaction of oleic acid and methanol. Results shown that the B-LaPW12O40/SiO2 has highest catalytic activity and stability: the conversion of oleic acid can be high to 93% when the molar ratio of methanol to oleic acid was 8:1, mass ratio of catalyst to reactants was 2%, reaction temperature was 65 ℃ and reaction time was 1 h; the conversion of oleic acid maintained 86.4% after B-LaPW12O40/SiO2 had been cycled six times. The high catalytic activity and stability of B-LaPW12O40/SiO2 can be explained as follows: a SiO2 network was formed from the hydrolytic action of Si(OC2H5)4 (TEOS) under acidic conditions via Sol-Gel process. The H+ of H3PW12O40 will bond with Si-OH in SiO2 network to form a (≡Si-OH2+)(H2PW12O40-) complex with strong electrostatic adsorption force, thus promoting the adsorption of La3+ on the surface of SiO2, greatly. As a result, the pore structure of H3PW12O40 will be blockaged, the grow up of H3PW12O40 particles in the roasting process also will be inhibited. In addition, SiO2 may be existed in the form of dry gel in the B-LaPW12O40/SiO2 catalyst and acted as carrier. It will be favorable for the improvent of the surface area of B-LaPW12O40/SiO2 since SiO2 has high surface area, so the surface area of B-LaPW12O40/SiO2 has increased from the 1.4 m2/g of H3PW12O40 to the 31.3 m2/g. And more, LaPW12O40/SiO2 has been determined from the Py-FTIR spectra of pyridine adsorption analysis, which is a Brönsted-Lewis solid acid. The formation of Lewis acid sites can help to reduce the deactivation of a solid acid catalyst: some H2O will be generated from the esterification reaction, and hydration will occur between Brönsted acid site and H2O, so the deactivation will occur. The formation of Lewis acid sites can be ascribed to the strong electrophilic action of La3+ after it has been bonded with (≡Si-OH2+)(H2PW12O40-) to form LaPW12O40/SiO2.
-
随着中国工业化进程不断加快,人们在消耗大量来自于化石资源能源(如石油、煤炭等)的同时,也对自然环境产生了严重的污染。石油和煤炭均属于不可再生资源,大量使用已经对中国的经济社会发展带来了巨大经济压力。除此之外,随着PM2.5、雾霾、汽车尾气等引起的环境污染问题不断出现,环境保护问题也越来越引起人们的关注。因此,发展和利用对环境无污染的可再生能源已成为中国当前迫切需要解决的一个重大课题。在众多可再生能源中,具有原料来源广泛和价格相对低廉等优势的生物质能源,已经获得了社会各界的广泛关注。
生物质能源中可直接作为汽车或其他动力装置的燃料使用的主要包括甲醇、乙醇和生物柴油。相比于甲醇和乙醇,由于生物柴油的能量密度与石化柴油接近,因而除开可被生物降解、无毒、不含硫、对环境无害,可达到美国“清洁空气法”所规定的健康影响检测要求等优点外,其还可满足无需对现有柴油机进行改造,可直接或按任意比例与石化柴油调配后在柴油机上使用的要求,是一种可用于替代石化柴油的理想清洁燃料。作为一种极具发展潜力的清洁燃料,全球生物柴油的产量在2016年达到了3.28×106 t,产量较高的国家依次为美国、巴西和德国,欧洲地区占所有生物柴油产量的43%左右,中国的产量约为3.36×104 t[1]。
按照当前生物柴油生产工艺的特点可知,原料成本占了生物柴油总生产成本的50%-85%。因此,原料成本一直是决定生物柴油价格的最主要因素。当前,生物柴油的主要生产国都根据各自国家所具有的植物油种植业的特点,选择了不同种类植物油来作为生物柴油的生产原料,如美国和欧盟等国家主要以菜籽油和大豆油为原料;巴西则主要使用蓖麻籽油为原料;印度、韩国、马来西亚等东南亚国家以棕榈油为原料。中国虽然是一个具有丰富植物资源且分布广泛优势的国家,可为生物柴油原料的选择提供巨大便利。但是,中国是人口大国,目前中国的植物油仍旧需要大量进口且食用优先,这决定了中国不可能利用大量的可食用植物油来作为生物柴油的生产原料。因此,欧美国家的生物柴油工业化发展模式不符合中国的国情。同时,由于中国是食用油消费大国,每年的食用油消费总量约为1.6×106 t。其中,大约有10%的食用油在使用后被废弃,产生了1.6×105 t废弃动植物油脂[2]。虽然废弃动植物油脂存在收集困难,预处理复杂等缺点,但具有回收价格低,可降低生物柴油生产原料成本的优点,如加以回收利用,无疑是一个巨大的廉价生物柴油原料来源,而且可以减少环境污染。因而,如能把废弃动植物油脂合理利用于生产生物柴油,可预见其将对中国的经济社会发展产生巨大的经济和环保效益。但是,废弃动植物油脂中的成分复杂,当以其为原料生产生物柴油时,对于一种能够在反应过程中保持高催化活性和稳定性的催化剂提出了非常苛刻的要求。
通过固体酸同时催化酯化与酯交换反应转化废弃动植物油脂为生物柴油,已经成为了经化学催化法合成生物柴油的新发展趋势。固体酸催化剂通常具有以下优势:易从产品中分离、对生产设备和管路阀门腐蚀小、对环境污染小、催化稳定性好[3]。其中,杂多酸作为一种具有高催化活性且对生产设备几乎无腐蚀的环境友好型固体酸催化剂,现已经被尝试用于同时催化酯化与酯交换反应转化废弃动植物油脂为生物柴油。目前,适合作为同时酯化与酯交换反应催化剂的主要是具有Keggin结构的12系列杂多酸(分子通式为HnAB12O40·xH2O),如十二硅钨酸、十二磷钨酸和十二磷钼酸等[4-7]。但是,表面反应的催化活性通常与表面酸性的大小密切相关,而杂多酸的比表面积较小(<10 m2/g)。因此,如何增大杂多酸的比表面积,显得非常关键。
当前的研究已经发现,不同抗衡离子(如铵根离子、钾离子、铯离子等)可与杂多酸的阴离子发生协同作用,加速杂多酸表面游离氧的迁移而对杂多酸的活性中心产生分散作用,且分散作用的效果随抗衡离子的直径增加而增强[8-10]。稀土元素离子通常具有较大的直径,因此,可选择稀土元素离子对杂多酸进行改性作用,以提高其比表面积和表面酸性,最终实现提高催化活性的目标。如在磷钼钒杂多酸中引入Ce4+改性作用后,当将其用于催化苯酚氧化羰基化合成碳酸二苯酯(DPC)时,发现催化剂具有了更高的催化活性,DPC的收率可达到6.7%[11]。又如在磷钼钒杂多酸中引入La3+改性作用后,当用于催化氧化甲基丙烯醛(MAL)为甲基丙烯酸(MAA)时,表现出了高催化活性,MAA的转化率可达到94.7%[12]。
综上可知,杂多酸的比表面积和表面酸性与其分子结构中担负抗衡离子作用的阳离子的直径密切相关。因而可通过改变其分子结构中的阳离子类型来解决其比表面积较小的缺陷,从而适合作为一种能同时高效催化酯化与酯交换反应而转化废弃动植物油脂为生物柴油的催化剂。由于Keggin结构的12系列杂多酸对酯化反应具有较高的催化活性,因而本研究以磷钨杂多酸(tungstophosphoric acid, H3PW12O40)为基体,并考虑到镧属于轻稀土,具有价格低廉的优点,且考虑到当前的研究较少报道稀土元素改性对提高H3PW12O40的比表面积和表面酸性的效果,是否与制备方法有一定的影响关系,而分别采用普通浸渍法、超声浸渍法和溶胶凝胶法,通过La3+改性H3PW12O40合成了三种固体酸催化剂A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40,并比较了以上不同方法制备而成的催化剂在用于催化以油酸和甲醇为反应物经酯化反应合成生物柴油时的催化活性和稳定性的差异。
1 实验部分
1.1 试剂和仪器
试剂:硝酸(65%, AR,西陇化工股份有限公司),磷钨酸(99%,AR,阿拉丁试剂有限公司),六水合硝酸镧(99.9%,AR,阿拉丁试剂有限公司),硅酸四乙酯(99%,GC,阿拉丁试剂有限公司),无水乙醇(99%,AR,衡阳市凯信化工试剂股份有限公司),曲拉通X-100(99.5%,CP, 国药集团化学试剂有限公司),甲醇(99%, AR,天津市大茂化学试剂厂),油酸(99%, AR,西陇化工股份有限公司),乙醚(99.5%,AR,西陇化工股份有限公司)和酚酞(99%,AR,天津市光复精细化工研究所)。
仪器:数控超声波清洗器(KQ-S00DE,昆山市超声仪器有限公司),集热式磁力加热搅拌器(DF-101S,金坛市白塔新宝仪器厂),电子天平(AR223CN,奥豪斯仪器有限公司),电热恒温干燥箱(DHG-9036A,上海精宏实验设备有限公司),循环水式真空泵(SHZ-D(Ⅲ),巩义市予华仪器有限责任公司),离心机(TGL-16C,上海安亭科学仪器厂),马弗炉(SXL-1200C,上海钜晶精密仪器制造有限公司)。
1.2 催化剂的制备
超声浸渍法:在50 mL烧杯中加入10 mL的0.1 mol/L硝酸、2.0 g磷钨酸和0.1 g六水合硝酸镧,搅拌均匀直至无明显沉淀出现;将混合液在超声波振荡仪中振荡处理30 min至溶液均匀,再机械搅拌作用5 min,静置24 h后,置于电热恒温干燥箱内,并设置温度为110 ℃进行干燥处理,直至将溶剂蒸干;将所得到的固体物质置于马弗炉中,在空气氛围中于300 ℃煅烧3 h,升温速率为5 ℃/min,冷却后用研钵研碎均匀,即得样品A-LaPW12O40。
溶胶-凝胶法:在50mL的烧杯中先依次加入硅酸四乙酯1.15 g、无水乙醇0.7 mL和0.1 mol/L的硝酸10 mL,再加入10 μm的曲拉通X-100;将以上物质混合后形成的溶液搅拌均匀,放入超声波振荡仪中振荡处理2 min;在超声处理后的溶液中加入2.0 g磷钨酸和0.1 g六水合硝酸镧,静置直至出现凝胶;将凝胶置于干燥箱中,在空气氛围中于110 ℃煅烧3 h,升温速率为2 ℃/min;将干凝胶用研钵研碎成细粉,即得样品B-LaPW12O40/SiO2。
常规浸渍法:在50 mL烧杯中依次加入10 mL的0.1 mol/L硝酸、2.0 g磷钨酸和0.1 g六水合硝酸镧,搅拌均匀直至无明显沉淀物质出现,静置24 h;置于电热恒温干燥箱内,在110 ℃下进行干燥处理,直至溶剂蒸干;将所得到的固体物质置于马弗炉中,在空气氛围中于300 ℃煅烧3 h,升温速率为5 ℃/min;用研钵研碎均匀,即得样品C-LaPW12O40。
1.3 催化剂的表征
通过X射线荧光光谱仪(PW2424, 荷兰帕纳科公司)分析H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂中元素的种类和含量。将测试样压片后,通过能量色散X荧光分析法进行分析,X射线源的管电压和管电流分别为50 kV和1 mA。采用物理吸附仪(ASAP 2020型,美国麦克仪器公司)测定了A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的比表面积、孔径、孔容和N2吸附-脱附等温线(196 ℃)。测试前试样在1×10-4 Pa和150 ℃的条件下抽真空预处理,液氮作吸附质,吸附温度为-196 ℃,通过Langmuir模型计算比表面积。通过TEM(Tecnai G2 F20,美国菲达康有限责任公司)对H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的形貌结构进行了观察。分析测试过程如下,将微量测试样溶解在无水乙醇中,并超声处理10 min,再把稀释后的样品置于带碳涂层的铜网上,待乙醇挥发完即可进行分析。加速电压200 kV,点分辨率0.24 nm。H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的晶型结构由XRD(EMPYREAN,荷兰帕纳科公司)分析,采用Cu Kα为辐射源,管电压40 kV,入射波长λ为0.154 44 nm,管电流40 mA,扫描速率为4 (°)/min。通过热重仪(TG/DTA6300,美国PE公司)分析了H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂在不同温度下失重的情况。测试条件为试样质量:6.527 mg,升温速率:20 ℃/min,气氛:O2,温度:25-900 ℃。通过傅里叶变换红外光谱仪(Magna-IR 750,美国尼高力公司),对A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂表面分子官能团进行了分析。测试过程如下:将2 mg粉末样品和经红外灯充分干燥后的200 mg KBr在研钵中研碎均匀,压片后进行分析。并结合吡啶吸附法,进一步对B-LaPW12O40/SiO2催化剂的酸性活性位类型和强度进行了分析。测试过程如下:将粉末样品压成直径为15 mm的自支撑片,质量10-20 mg。升温至350 ℃,并抽真空至10-3 Pa,保持1 h,脱除样品中的气体分子。降至室温,吸附吡啶0.5 h,吸附平衡后分别升温至200、350 ℃,脱附0.5 h,冷却至室温后扫描1 400-1 700 cm-1,得到样品经过200、350 ℃脱附的吡啶吸附红外光谱。扣除本底后,得到不同温度下的吡啶吸附红外光谱图。在程序升温仪(AutoChem Ⅱ 2920,美国麦克公司)上,通过NH3程序升温脱附法(NH3-TPD)分析了催化剂的酸性活性位的强度,测量温度范围为0-800 ℃。通过多功能成像电子能谱仪XPS(Thermo Scientific Escalab 250Xi,美国赛默飞世科技公司)对B-LaPW12O40/SiO2催化剂的价键类型进行了分析,采用单色Al Kα (hv =1 486.6 eV),功率150 W,500 μm束斑。
1.4 催化剂活性及稳定性
通过油酸与甲醇酯化反应,对A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的活性进行了比较研究。实验条件如下:在250 mL三口烧瓶中加入物质的量之比为8:1的甲醇和油酸,并按油酸与甲醇总质量的2%分别加入A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂,将反应温度设置为65 ℃,并在此温度下回流反应8 h。通过酸值法测量分析油酸的转化率,酸值测定按GB 5530-85《植物油脂检验酸值测定法》中介绍的测定方法进行。对样品进行酸值测定前的预处理步骤如下:每隔1 h取一次样(取1.5 mL样品,取完样后再加入同体积同物质的量比的甲醇和油酸),将测试样放入干燥箱中,在80 ℃下加热30 min以蒸出测试样中残余的甲醇(甲醇沸点为64.7 ℃),之后将测试样进行离心作用以除去残余的催化剂,以得到纯度较高的测试样来进行酸值分析而避免误差。对催化剂的稳定性进行分析时,采用乙醚萃取法将催化剂从反应体系中分离出来并循环使用六次,具体如下:将包含催化剂的反应物置于分液漏斗中,待反应物冷却后,加入乙醚进行萃取,收集上层,将醚进行蒸发,即析出催化剂。
2 结果与讨论
对催化剂中的元素种类和含量进行分析,将有助于研究者结合催化剂稳定性及活性的数据确定催化剂中的元素种类和含量在反应过程中所产生的作用。因此,对H3PW12O40、A-LaPW12O40和B-LaPW12O40/SiO2、C-LaPW12O40催化剂中的元素种类和含量进行了分析,结果见表 1。
 表 1
催化剂中各元素的含量
Table 1.
Atomic percentages of different elements of four catalysts
表 1
催化剂中各元素的含量
Table 1.
Atomic percentages of different elements of four catalysts

由表 1可知,A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂中都含有La元素。其中,A-LaPW12O40和B-LaPW12O40/SiO2中的La含量几乎相同,分别为2.248%和2.244%,而C-LaPW12O40中的La含量仅为1.430%,由此表明超声波和溶胶-凝胶法均有助于La3+进入H3PW12O40的骨架结构。
当前的一些研究已经发现稀土元素离子由于直径较大而可有效提高杂多酸的分散度,但较少报道该效果是否同时还与制备方法有一定的影响关系。因此,为了解La3+改性H3PW12O40方法的差异对制备而成的催化剂的比表面积、孔容和孔径是否会产生较大的影响,而对H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的比表面积、孔容、孔径进行了测量分析,结果见表 2。
表 2
催化剂的比表面积、孔容和孔径
Table 2.
Surface area, pore volume and pore size of catalysts

由表 2可知,B-LaPW12O40/SiO2的比表面积明显大于H3PW12O40,而A-LaPW12O40和C-LaPW12O40的比表面积和孔径均小于H3PW12O40。由此可得出以下结论,通过在H3PW12O40中引入La3+来提高其比表面积具有较好的效果,但该效果与所采用的改性方法密切相关,溶胶-凝胶法是一种有效的方法。这可归因于在溶胶-凝胶法中使用了TEOS,而使合成的B-LaPW12O40/SiO2催化剂中含有较多的Si元素,其可与H3PW12O40骨架上的氧原子配位桥联形成O-Si-O化学键,随着La3+引入H3PW12O40的骨架后,La3+将随机吸附在SiO2表面,由于La3+产生的强静电吸附力而导致H3PW12O40的孔道结构发生部分堵塞,这在一定程度上抑制了H3PW12O40颗粒在焙烧过程中的进一步聚集长大。除此之外,SiO2经110 ℃干燥处理后,可能将以干凝胶状态存在于B-LaPW12O40/SiO2催化剂中,并起到载体的作用,由于SiO2凝胶的高比表面积,而使B-LaPW12O40/SiO2具有了较大的比表面积。图 1为H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的透射电镜照片。
 图 1
A-LaPW12O40、B-LaPW12O40/SiO2、C-LaPW12O40和H3PW12O40催化剂的透射电镜照片
Figure 1.
TEM images of A-LaPW12O40, B-LaPW12O40/SiO2, C-LaPW12O40 and H3PW12O40
图 1
A-LaPW12O40、B-LaPW12O40/SiO2、C-LaPW12O40和H3PW12O40催化剂的透射电镜照片
Figure 1.
TEM images of A-LaPW12O40, B-LaPW12O40/SiO2, C-LaPW12O40 and H3PW12O40
由图 1可知,H3PW12O40(TPA)分布均匀且呈笼状结构。通过超声浸渍法合成的A-LaPW12O40保持了笼状结构,与H3PW12O40未经La3+改性之前的密集分布状态相比,活性中心的分散性已经得到了明显的改善,这说明了La3+可有效提高杂多酸的分散度。然而,由表 2可知,A-LaPW12O40的比表面积小于H3PW12O40。以上说明了超声辅助浸渍法无法同时兼顾增大H3PW12O40比表面积和提高其分散度的作用效果。通过溶胶-凝胶法合成得到的B-LaPW12O40/SiO2,兼顾了增大H3PW12O40比表面积和提高其分散度的作用效果,这可归因于作为载体的SiO2,具有强分散能力,可使活性组分高度分散。同样,由表 2还可知,通过常规浸渍法合成得到的C-LaPW12O40的比表面积小于H3PW12O40,且活性中心的分散性没有得到明显的改善,这是由于常规浸渍无法使La3+发生选择性吸附作用,导致其在经过经热处理后, 以大小不一的金属颗粒形式出现在载体表面。采用溶胶凝胶法对H3PW12O40进行La3+改性作用,可同时增大其比表面积和提高其分散度。图 2为H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的XRD分析谱图。
 图 2
催化剂的XRD谱图
Figure 2.
XRD patterns of four catalysts
图 2
催化剂的XRD谱图
Figure 2.
XRD patterns of four catalysts
由图 2可知,H3PW12O40在10.3°、25.4°、34.6°出现了对应于Keggin结构的(110)、(222)、(332) 晶面衍射峰,峰位和峰高符合立方晶系H3PW12O40·6H2O (JCPDS 00-050-0304)。与H3PW12O40相比,除了有些峰位发生了微小的偏移之外,A-LaPW12O40和C-LaPW12O40的XRD谱图上的主峰与H3PW12O40基本一致,由此表明通过常规浸渍和超声浸渍法对H3PW12O40进行La3+改性作用后,并没有破坏H3PW12O40的Keggin结构。而比较H3PW12O40和B-LaPW12O40/SiO2的XRD谱图可发现两者的主峰有较大的差异。B-LaPW12O40/SiO2的主峰向低角度方向发生了偏移,分别在8.0°、20.0°、24.3°处出现了与三斜晶系H3PW12O40 (JCPDS 00-050-0656) 非常匹配的晶面衍射峰,该晶型结构的变化可归因于采用溶胶凝胶合成B-LaPW12O40/SiO2时,在溶胶凝胶的转化过程中,TEOS会在酸性条件下水解为硅醇基,并质子化后成为杂多阴离子的抗衡离子,最终生成(Si-OH2+)(H2PW12O40-)络合物,而导致H3PW12O40的Keggin的结构发生一定程度形变[13]。图 3为H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的TG谱图。
 图 3
催化剂的TG、DTA、DTG曲线
Figure 3.
TG, DTA, DTG analysis of four catalysts
图 3
催化剂的TG、DTA、DTG曲线
Figure 3.
TG, DTA, DTG analysis of four catalysts
由图 3可知,H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂都在低于200 ℃有个明显的失重峰,对应于物理或化学吸附水的失重。经对比H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2的失重曲线后,可发现三者在温度高于200 ℃的失重趋势图相似,均在低于200 ℃发生较大的失重,之后趋于稳定。经比较失重量可发现B-LaPW12O40/SiO2催化剂中的含水量最大,当温度到达200 ℃时其已失重了28%,这可能是由于络合生成(Si-OH2+)(H2PW12O40-)的过程中,有部分水进入了H3PW12O40的结构中。图 4为H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的红外光谱分析图。
 图 4
催化剂的红外光谱谱图
Figure 4.
Infrared spectra of four catalysts
图 4
催化剂的红外光谱谱图
Figure 4.
Infrared spectra of four catalysts
由图 4可知,H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的红外光谱图无明显差异,A-LaPW12O40和B-LaPW12O40/SiO2、C-LaPW12O40催化剂都保留了Keggin结构的四个特征峰,分别为1 080 cm-1处的P-O伸缩振动吸收峰,977 cm-1处的W=O振动吸收峰,895 cm-1处的W-Oa-W共点振动吸收吸收峰和808 cm-1处的W-Ob-W共棱振动吸收峰[14]。相比于H3PW12O40,A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂中的P-O键均向高波数方向移动了几个单位,这是由于La-O键在1 100 cm-1处有强振动峰,而易与P-O键在1 080 cm-1处的特征峰发生叠加所致。
图 5为H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的N2吸附-脱附等温线图。
 图 5
催化剂的N2吸附-脱附等温线
Figure 5.
N2 adsorption-desorption isotherms of four catalysts
图 5
催化剂的N2吸附-脱附等温线
Figure 5.
N2 adsorption-desorption isotherms of four catalysts
由图 5可知,H3PW12O40、A-LaPW12O40和C-LaPW12O40的N2吸附-脱附等温线相似,均随着p/p0增大呈现出稍微的上升趋势,属于Type Ⅱ型吸附等温线[15],即为非多孔性固体表面或大孔固体上发生的单层到多层可逆吸附过程,这也表明了A-LaPW12O40、C-LaPW12O40和H3PW12O40为介孔材料,与孔径的测量结果一致。由此也证明了A-LaPW12O40、C-LaPW12O40和H3PW12O40在作为催化剂使用时,发生的是单分子层吸附。而B-LaPW12O40/SiO2的N2吸附-脱附等温线没有闭合,这可能是由于其孔径值较小,接近于微孔材料,且在经过高压吸附之后,作为载体的SiO2的孔结构发生了坍塌。图 6为H3PW12O40、A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的氨程序升温脱附分析图。
 图 6
催化剂的氨程序升温脱附曲线
Figure 6.
NH3 temperture-programmed desorption profiles of four catalysts
图 6
催化剂的氨程序升温脱附曲线
Figure 6.
NH3 temperture-programmed desorption profiles of four catalysts
由图 6可知,H3PW12O40、A-LaPW12O40均只在600-700 ℃出现了一个脱附峰,且A-LaPW12O40的脱附峰强度要弱于H3PW12O40;C-LaPW12O40在100-250 ℃出现了一个脱附峰;B-LaPW12O40/SiO2则分别在100-250 ℃和600-700 ℃各出现了一个脱附峰。以上表明,H3PW12O40和A-LaPW12O40含有的酸位主要是强酸位,且H3PW12O40的强酸位浓度高于A-LaPW12O40,这可能是由于H3PW12O40中的H+被La3+取代后,减弱了其酸性;C-LaPW12O40则以弱酸位为主,这可能是由于La3+与H3PW12O40上的氧原子配位桥联时发生了团聚,使H3PW12O40的酸性活性位点之间发生了相互屏蔽,而导致了其酸性明显减弱;B-LaPW12O40/SiO2则分别含有强酸位和弱酸位,弱酸位的出现同样可归因于H3PW12O40中的H+被La3+取代后而减弱了其酸性,强酸位的出现则可归因于具有强静电吸附作用的(≡Si-OH2+)(H2PW12O40-)络合物的形成。
基于以上对H3PW12O40、A-LaPW12O40和B-LaPW12O40/SiO2、C-LaPW12O40催化剂所进行的物理化学性质分析结果,进而对以上催化剂在油酸与甲醇酯化反应中的催化活性进行了比较研究,反应条件如下:甲醇与油酸的物质的量比为8:1,催化剂用量为反应物总质量的2%,反应温度为65 ℃,反应时间为8 h,反应结果见图 7。
 图 7
催化剂的活性对比
Figure 7.
Comparison of catalytic activity
图 7
催化剂的活性对比
Figure 7.
Comparison of catalytic activity
由图 7可知,相比于H3PW12O40,A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂均具有更高的催化活性,其中,活性最高的是B-LaPW12O40/SiO2催化剂。以H3PW12O40为催化剂,反应8 h后油酸的转化率为86%。而以B-LaPW12O40/SiO2为催化剂时,反应1 h后油酸的转化率即高达93%,以上可归因于通过溶胶-凝胶法合成得到的B-LaPW12O40/SiO2催化剂具有较大的比表面积和表面酸性。为了判断B-LaPW12O40/SiO2催化剂与之前文献报道的其他类型同样通过催化油酸与甲醇酯化反应合成生物柴油的固体酸催化剂的活性大小,而进行了催化活性比较分析,结果见表 3。
表 3
不同固体酸在催化油酸和甲醇酯化反应合成生物柴油时的催化活性比较
Table 3.
Comparison of activity of different solid acids for the synthesis of biodiesel from esterification of oleci acid and methanol

在催化活性比较的基础上,进而对H3PW12O40,A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40催化剂的催化稳定性进行了比较研究,反应条件如下,甲醇与油酸的物质的量比为8:1,反应温度为65 ℃,催化剂占反应物的质量比为2%,反应时间为1 h,反应结果见图 8。
 图 8
催化剂的稳定性
Figure 8.
Comparison of catalytic stability
图 8
催化剂的稳定性
Figure 8.
Comparison of catalytic stability
由图 8可知,相比于H3PW12O40、A-LaPW12O40和C-LaPW12O40催化剂,催化稳定性最高的是B-LaPW12O40/SiO2催化剂,经过六次循环使用后,油酸的转化率仍可以到86.4%,由此说明稀土元素La改性对提高H3PW12O40的比表面积和表面酸性的效果与制备方法有较高的影响关系,溶胶凝胶法是一种适合的制备方法,B-LaPW12O40/SiO2适合作为通过油酸与甲醇酯化反应合成生物柴油的催化剂。
为了进一步了解B-LaPW12O40/SiO2催化剂的酸性活性位的类型,采用吡啶红外吸附法对其进行了分析,结果见图 9。
 图 9
B-LaPW12O40/SiO2催化剂的吡啶红外吸附谱图
Figure 9.
Pyridine adsorption Py-FTIR spectrum of B-LaPW12O40/SiO2 catalyst
图 9
B-LaPW12O40/SiO2催化剂的吡啶红外吸附谱图
Figure 9.
Pyridine adsorption Py-FTIR spectrum of B-LaPW12O40/SiO2 catalyst
由图 9可知,在波数为1 540和1 450 cm-1处均出现了特征峰,分属于Brönsted酸位和Lewis酸位所对应的特征峰[20],由此表明,B-LaPW12O40/SiO2是一种Brönsted酸位和Lewis酸位同时存在的酸性催化剂。H3PW12O40是一种酸强度较为均一的纯质子酸,即Brönsted酸[21]。而B-LaPW12O40/SiO2中出现的Lewis酸位,可能是由于(Si-OH2+)(H2PW12O40-)进一步与吸附在其表面的La3+形成了(La3+-Si-OH2+)(H2PW12O40-),由于La3+的强吸电子作用而有助于催化剂中Lewis酸位的形成,从而增加了Lewis酸位的含量。由于Brönsted酸位易与酯化反应过程中生成的水发生水合反应而失活,因而Lewis酸位的形成有助于提高催化剂的稳定性。为了解B-LaPW12O40/SiO2催化剂中所含有的价键类型,采用X射线光电子能谱法对其进行了分析,结果见图 10。
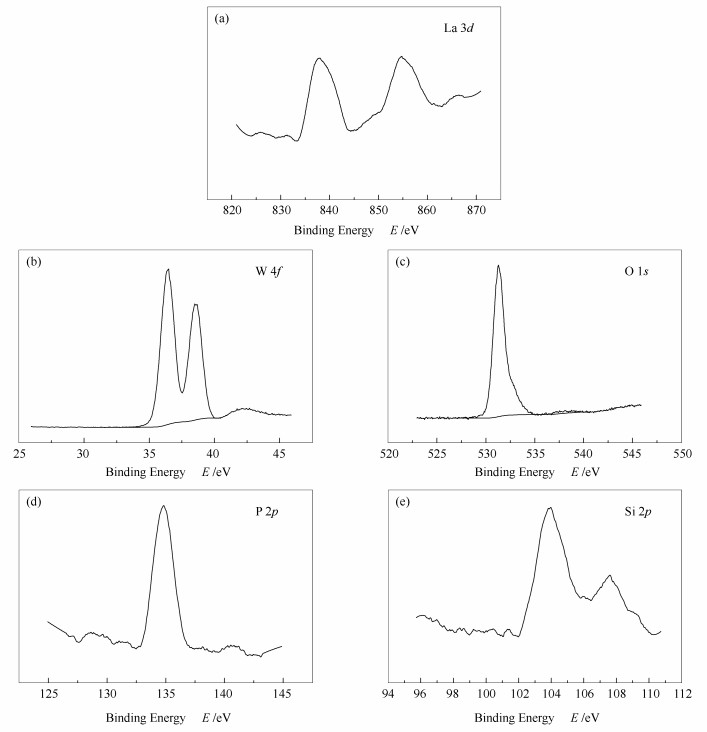 图 10
催化剂B的X射线光电子能谱图
Figure 10.
Catalyst B X-ray photoelectron spectroscopy of La 3d(a), W 4f(b), O 1s(c), P 2p(d), Si 2p(e)
图 10
催化剂B的X射线光电子能谱图
Figure 10.
Catalyst B X-ray photoelectron spectroscopy of La 3d(a), W 4f(b), O 1s(c), P 2p(d), Si 2p(e)
图 10(a)为B-LaPW12O40/SiO2催化剂的La 3d光电子吸收峰,分别为835.5 eV的La 3d5/2和856.4 eV的La 3d3/2的光电子吸收峰,而当La的4f轨道未受到任何化学环境影响时,La 3d5/2和La 3d3/2的光电子吸收峰将分别出现在833.9 eV和850.0 eV处[22]。由此可知,La 3d5/2和La 3d3/2的结合能均向高能方向发生了偏移,这是由于La3+与(≡Si-OH2+)(H2PW12O40-)发生了强相互作用,两者之间有电子转移而出现的结果。图 10(b)为W 4f能谱,分别在36.5和38.6 eV处出现了对应于W 4f7/2和W 4f5/2的光电子吸收峰,而纯磷钨酸的W 4f7/2和W 4f5/2光电子吸收峰分别位于35.8和37.8 eV [21],因而W 4f7/2和W 4f5/2结合能同样向高能方向发生了偏移,结合能的提高应该是在溶胶凝胶的转化过程中,H3PW12O40由于存在有不同程度的氧缺位,使得部分W6+被还原为W5+。且在图 10(c)中O 1s只有533.1 eV处的光电子吸收峰,并结合图 10(d) P 2p能谱在135 eV处出现的峰,以及图 10(e) Si 2p在104和108 eV处出现的光电子吸收峰可知该催化剂中含有La-O键、W-O-W键和O-P-O键[21, 22]。
3 结论
研究以磷钨杂多酸(tungstophosphoric acid, H3PW12O40)为基体,分别通过普通浸渍法、溶胶凝胶法和超声浸渍法进行了La3+改性作用,合成了三种固体酸催化剂A-LaPW12O40、B-LaPW12O40/SiO2和C-LaPW12O40,并比较了以上催化剂在用于催化以油酸和甲醇为反应物经酯化反应合成生物柴油时的催化活性的差异。结果表明,B-LaPW12O40/SiO2具有最高催化活性和稳定性,以其为催化剂,当甲醇与油酸的物质的量比为8:1,催化剂用量为反应物总质量的2%,反应温度为65 ℃,反应1 h后,油酸的转化率即高达93%。B-LaPW12O40/SiO2经过六次循环使用后,在与催化活性研究相同的实验条件下,反应1 h后的油酸转化率仍可达86.4%。B-LaPW12O40/SiO2的高催化活性和稳定性可归因于在采用溶胶凝胶法合成其时,在溶胶凝胶的转化过程中,作为硅源材料的四乙氧基硅(TEOS)易在酸性条件下发生水解作用形成SiO2网络,H3PW12O40中的H+会与SiO2网络中的硅醇键结合并形成具有强静电吸附力的(≡Si-OH2+)(H2PW12O40-)络合物,这将促进La3+在SiO2表面的吸附而堵塞H3PW12O40的孔道结构,并抑制H3PW12O40颗粒在焙烧过程中的进一步聚集长大。SiO2将作为载体并以干凝胶状态存在于B-LaPW12O40/SiO2催化剂中,由于SiO2凝胶的高比表面积而使而使B-LaPW12O40/SiO2具有较大的比表面积。并且,(≡Si-OH2+)(H2PW12O40-)将进一步与吸附在其表面的La3+发生键结合作用,由于La3+具有强吸电子作用,该作用将有助于Lewis酸位的形成而增加其含量。由于Brönsted酸位易与酯化反应过程中生成的水发生水合反应而失活,因而Lewis酸位的形成有助于减少催化剂的失活现象发生。因此,当通过油酸与甲醇酯化反应合成生物柴油时,B-LaPW12O40/SiO2是一种具有高催化活性和稳定性的Brönsted-Lewis酸型固体酸催化剂。
-
-
[1]
HAJJARI M, TABATABAEI M, AGHBASHLO M, GHANAVATI H. A review on the prospects of sustainable biodiesel production: A global scenario with an emphasis on waste-oil biodiesel utilization[J]. Renew Sustain Energy Rev, 2017, 72: 445-464. doi: 10.1016/j.rser.2017.01.034
-
[2]
XU Y J, LI G X, SUN Z Y. Development of biodiesel industry in China: Upon the terms of production and consumption[J]. Renew Sustain Energy Rev, 2016, 54: 318-330. doi: 10.1016/j.rser.2015.10.035
-
[3]
VHAD M R, MARCHETTI J M. A review on recent advancement in catalytic materials for biodiesel production[J]. Sust Energ Rev, 2015, 50: 696-718. doi: 10.1016/j.rser.2015.05.038
-
[4]
SINGH S, PATEL A. Selective green esterification and oxidation of glycerol over 12-tungstophosphoric acid anchored to MCM-48[J]. Ind Eng Chem Res, 2014, 53: 14592-14600. doi: 10.1021/ie5026858
-
[5]
SERT E, ATALAY F S. Esterification of acrylic acid with different alcohols catalyzed by zirconia supported tungstophosphoric acid[J]. Ind Eng Chem Res, 2012, 51: 6666-6671. doi: 10.1021/ie202609f
-
[6]
LI L X, LIU B Y, WU Z W, YUAN X, LUO H A. Preparation of Keggin-type mono-lacunary phosphotungstic-ammonium salt and its catalytic performance in ammoximation of cyclohexanone[J]. Chem Eng J, 2015, 280: 670-676. doi: 10.1016/j.cej.2015.06.048
-
[7]
CAICEDO A M E, RENGIFO-HERRERA J A, FLORIAN P, BLANCO M N, ROMANELLI G P, PIZZIO L R. Valorization of biomass derivatives: Keggin heteropolyacids supported on titania as catalysts in the suitable synthesis of 2-phenoxyethyl-2-furoate[J]. J Mol Catal A: Chem, 2016, 425: 266-274. doi: 10.1016/j.molcata.2016.10.024
-
[8]
ZHANG X Y, ZHANG D, SUN Z, XUE L F, WANG X H, JIANG Z J. Highly efficient preparation of HMF from cellulose using temperature-responsive heteropolyacid catalysts in cascade reaction[J]. Appl Catal B: Environ, 2016, 196: 50-56. doi: 10.1016/j.apcatb.2016.05.019
-
[9]
WU X Z, LIU Y T, LIU R, WANG L L, LU Y B, XIA X N. Hydroxyalkylation of phenol to bisphenol F over heteropolyacid catalysts: The effect of catalyst acid strength on isomer distribution and kinetics[J]. J Colloid Interf Sci, 2016, 481: 75-81. doi: 10.1016/j.jcis.2016.07.043
-
[10]
SHI H X, ZHANG T Y, AN T C, LI B, WANG X. Enhancement of photocatalytic activity of nano-scale TiO2, particles co-doped by rare earth elements and heteropolyacids[J]. J Colloid Interf Sci, 2012, 380: 121-127. doi: 10.1016/j.jcis.2012.04.069
-
[11]
戈军伟, 杜治平, 袁华, 杨小俊, 吴元欣. Keggin型磷钼矾杂多化合物在氧化羰基化合成碳酸二苯酯中的应用[J]. 应用化工, 2009,38,(1): 19-22. GE Jun-wei, DU Zhi-ping, YUAN Hua, YANG Xiao-jun, WU Yan-xin. Application of Keggin type molybdovanadophosphoric compounds in synthesis of diphenyl carbonate by oxidative carbonylation with phenol[J]. Appl Chem Ind, 2009, 38(1): 19-22.
-
[12]
郭晓俊, 黄崇品. 反荷离子及制备方法对Keggin型杂多化合物结构和性质的影响[J]. 石油化工, 2008,37,(3): 216-221. GUO Xiao-jun, HUANG Chong-pin. Effects of counter-ions and preparation methods on structures and properties of Keggin-type heteropoly compounds[J]. Petrochem Technol, 2008, 37(3): 216-221.
-
[13]
MATTOS F C G D, SOUZA J A D S D, COTRIM A B D A, MACEDO J L, DIAS J A, DIAS S C L, GHESTI G F. Lewis acid/surfactant rare earth trisdodecylsulfate catalysts for biodiesel production from waste cooking oil[J]. Appl Catal A: Gel, 2012, 423/424(8): 1-6.
-
[14]
MARCI G, GARCIA-LOPEZ E I, POMILLA F R, LIOTTA L F, PALMISANO L. Enhanced (photo) catalytic activity of Wells-Dawson (H6P2W18O62) in comparison to Keggin (H3PW12O40) heteropolyacids for 2-propanol dehydration in gas-solid regime[J]. Appl Catal A: Gen, 2016, 528: 113-122. doi: 10.1016/j.apcata.2016.10.002
-
[15]
WANG Z, FAN Y, Li Y W, QU F R, WU D Y, KONG H N. Synthesis of zeolite/hydrous lanthanum oxide composite from coal fly ash for efficient phosphate removal from lake water[J]. Micropor Mesopor Mat, 2016, 222: 226-234. doi: 10.1016/j.micromeso.2015.10.028
-
[16]
ZHANG Y, WONG W T, YUNG K F. Biodiesel production via, esterification of oleic acid catalyzed by chlorosulfonic acid modified zirconia[J]. Appl Energy, 2014, 116(1): 191-198.
-
[17]
SANTOS J S, DIAS J A, DIAS S C L, DE MACEDO J L, GARCIA F A C, ALMEIDA L S, DE CAVALHO E N C B. Acidic characterization and activity of (NH4)xCs2.5-xH0.5PW12O40 catalysts in the esterification reaction of oleic acid with ethanol[J]. Appl Catal A: Gen, 2012, 443/444: 33-39. doi: 10.1016/j.apcata.2012.07.013
-
[18]
MORENO J I, JAIMES R, GOMEZ R, GOMEZ M E N. Evaluation of sulfated tin oxides in the esterification reaction of free fatty acids[J]. Catal Today, 2011, 172(1): 34-40. doi: 10.1016/j.cattod.2011.03.052
-
[19]
JUNIOR C A R M, ALBURQUERQUE C E R, CARNEIRO J S A, DARIVA C, FORTUNY M, SANTOS A F, EGUES S M S, RAMOS A L. Solid-acid-catalyzed esterification of oleic acid assisted by microwave heating[J]. Ind Eng Chem Res, 2010, 49(23): 12135-12139. doi: 10.1021/ie100501d
-
[20]
CAMPOSECO R, CASTILLO S, MEJIA-CENTENO I, NAVARRETE J, RODRIGUEZ-GONZALEZ V. Behavior of lewis and brönsted surface acidity featured by Ag, Au, Ce, La, Fe, Mn, Pd, Pt, V and W decorated on protonated titanate nanotubes[J]. Micropor Mesopor Mat, 2016, 236: 235-243. doi: 10.1016/j.micromeso.2016.08.033
-
[21]
JALIL P A, FAIZ M, TABET N, HAMDAN N M, HUSSAIN Z. A study of the stability of tungstophosphoric acid, H3PW12O40, using synchrotron XPS, XANES, hexane cracking, XRD, and IR spectroscopy[J]. J Catal, 2002, 217: 292-297.
-
[22]
ZHANG J Q, WONG H, KAKUSHIMA K, IWAI H. XPS study on the effects of thermal annealing on CeO2/La2O3 stacked gate dielectrics[J]. Thin Solid Films, 2016, 600: 30-35. doi: 10.1016/j.tsf.2016.01.001
-
[1]
-

图 1 A-LaPW12O40、B-LaPW12O40/SiO2、C-LaPW12O40和H3PW12O40催化剂的透射电镜照片
Figure 1 TEM images of A-LaPW12O40, B-LaPW12O40/SiO2, C-LaPW12O40 and H3PW12O40

图 6 催化剂的氨程序升温脱附曲线
Figure 6 NH3 temperture-programmed desorption profiles of four catalysts

图 7 催化剂的活性对比
Figure 7 Comparison of catalytic activity
the molar ratio of methanol to oleic acid was 8:1, mass ratio of catalyst to reactants was 2%, reaction temperature was 65 ℃ and reaction time was 8 h

图 8 催化剂的稳定性
Figure 8 Comparison of catalytic stability
the molar ratio of methanol to oleic acid was 8:1, reaction temperature was 65 ℃, mass ratio of catalyst to reactants was 2% and reaction time was 1 h

图 9 B-LaPW12O40/SiO2催化剂的吡啶红外吸附谱图
Figure 9 Pyridine adsorption Py-FTIR spectrum of B-LaPW12O40/SiO2 catalyst

图 10 催化剂B的X射线光电子能谱图
Figure 10 Catalyst B X-ray photoelectron spectroscopy of La 3d(a), W 4f(b), O 1s(c), P 2p(d), Si 2p(e)
表 3 不同固体酸在催化油酸和甲醇酯化反应合成生物柴油时的催化活性比较
Table 3. Comparison of activity of different solid acids for the synthesis of biodiesel from esterification of oleci acid and methanol
 下载: 导出CSV
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1705
- HTML全文浏览量: 311
 下载:
下载:
