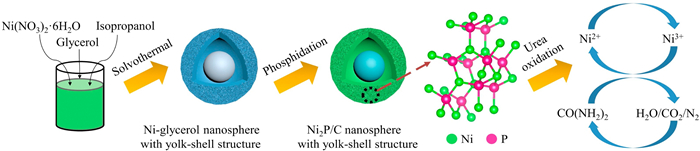
Scheme 1.
Schematic diagram of the preparation of a yolk-shell nanostructural catalyst of Ni2P/C.

Hydrogen production from natural resources and industrial waste water has been considered as one of the efficient routes to alleviate the serious energy crisis and ever-growing environmental issues. Fuel cells and the corresponding energy conversion techniques are recognized as the most potential strategies for hydrogen production due to the safety and efficiency in the transport and storage of hydrogen. In this regard, the methanol fuel cells (MFCs), ethanol fuel cells (EFCs), urea fuel cells (UFCs) and other devices with the low molecular weight organic species served as hydrogen rich fuel have gathered much attention [1-3]. Very recently, the developments of the urea electrochemical oxidation reaction deepen the insight of sustainable method to produce hydrogen since urea can be utilized as the promising fuels in direct urea fuel cell on an industrial scale by virtue of its non-toxicity, non-flammable, affordability nature [4, 5]. On the other hand, urea is also the common and abundant contaminant originated from human/animal urine, fertilizer industries and other wastes. The urea in the wastewater would convert to ammonia under natural conditions, and continue being oxidized to the next hydrolysed pollutants (e.g., nitrites, nitrate and nitric oxides) [6-8]. Hence, harmful environmental issues caused by the generated toxic and hazardous materials from urea also can be successfully treated.
Generally, urea electrolysis (anode: CO(NH2)2 + 6OH− → N2 + 5H2O+CO2 + 6e−, cathode: 6H2O + 6e− → 3H2 + 6OH−) exhibits the obvious advantages over the water splitting (anode: oxygen evolution reaction, cathode: hydrogen evolution reaction) in anodic reaction during hydrogen production, since the lower theoretical dynamic potential (0.37 V vs. 1.23 V) is required [9, 10]. However, the anodic urea oxidation reaction undergoes sluggish kinetics due to the 6e− transfer process, limiting the overall performance of urea electrolysis [11, 12]. Despite the recent remarkable capacities towards the urea oxidation reported for the precious metal based electrocatalysts (i.e., Rh, Ir and Pd), the scarcity and high cost of noble metals make it impossible for large scale applications [13-15]. So far, non-previous metal based catalysts, especially the nickel-based materials, have emerged as more popular and highly active urea oxidation reaction (UOR) catalysts. For example, Ni(OH)2, NiSe2, NiMoO4, NiLaO3 have been widely investigated in previous literatures [16-19].
Among the nickel-based materials, Ni2P has been widely investigated in the fields of electrochemistry, such as oxygen evolution reaction [20, 21], hydrogen evolution reaction [21, 22], supercapacitor [22, 23] and hydrodeoxygenation [24, 25], owning to its abundant availability, low cost, high activity and excellent stability. In case of UOR, it is well acknowledged that nickel phosphides are feasibly oxidized to nickel oxyhydroxide during the UOR process, serving as the essential active sites for catalyzing the urea oxidation. Nevertheless, the conductivity of electrode materials evidently decay after the formation of nickel oxyhydroxide, endowing the UOR process gradually deteriorated [26, 27]. In this regard, the nanostructure regulation via carbon coating is suggested to be one of the most efficient methods to improve the conductivity of the electrode material without changing the intrinsic catalytic mechanism [27, 28]. For instance, nickel hydroxide-carbon nanotubes composites (Ni(OH)2-CNTs) prepared via one-pot hydrothermal route exhibit the remarkable electrochemical urea oxidation performance with a maximal peak current density of 98.5 mA/cm2, which can be attributed to the unique lamellar CNTs conductive structure and the high trivalent Ni species contents [29]. Furthermore, it is reported that two-dimensional Ni/NiO nanosheets coated with ultrathin N-doped carbon layers are well designed, containing numerous Schottky heterointerfaces. The catalyst illustrates the potential of 1.35 V at 10 mA/cm2 for UOR, due to the feasible cleavage of urea molecules after self-driven charge redistribution at the heterointerface [30]. Inspired by these factors, coupling the active Ni2P with conductive carbon materials, together with merits of exposure of many active sites is highly desirable yet challenging.
Herein, we report one kind of novel york-shell-structural Ni2P/C nanosphere hybrids (termed as Ni2P/C-YS) working as the UOR catalyst. Owing to the optimized york-shell morphology, more active sites of the catalyst can be feasibly realized, giving rise to the advanced electrochemical activity towards the urea electro-oxidation. Meanwhile, the excellent performance also can be attributed to the improved electronic conductivity after introducing the coated carbon, which obviously optimizes the electron transfer channels and accelerates the catalytic kinetics. The as-prepared Ni2P/C-YS-350 nanocomposite exhibits the outstanding UOR catalytic capacity with a low potential of 1.336 V to drive a current density of 50 mA/cm2 and a low Tafel slope of 10.1 mV/dec in alkaline electrolyte and the superior durability with negligible potential decay after 23 h, highlighting the important value of their future application in the urea fuel cells using wastewater.
In a typical synthesis route of Ni-glycerate (Ni-gly) yolk-shell nanospheres, 0.7 mmol of Ni(NO3)2·6H2O and 10 mL of glycerol were added into a 60 mL isopropanol. After stirring for 5 min to form a homogeneous solution, the mixed solution together with the addition of 2 mL deionized (DI) water was transferred to a 100 mL Teflonlined stainless steel autoclave, and kept in an oven at 200 ℃ for 12 h. After naturally cooling to room temperature, the precipitate was separated by filtration with a 0.22 μm Nylon organic as the filter membrane, washed several times with DI water and ethanol. The Ni-gly precursor was obtained after dried at 60 ℃ overnight.
The Ni-gly precursor (100 mg) was then transfers into the Ni2P/C yolk-shell nanospheres by PH3 decomposed from sodium hypophosphite monohydrate at the temperature of 350 ℃ in an argon atmosphere for 2 h with a slow rate of 2 ℃/min in tube furnace. The final black powder product was collected as Ni2P/C (Ni2P/C-YS-350). For comparison, a diversity of yolk-shell Ni2P/C samples (Ni2P/C-YS-250, Ni2P/C-YS-300, Ni2P/C-YS-400, Ni2P/C-YS-450) were obtained under the phosphidation temperature of 250, 300, 400 and 450 ℃.
The crystal structures of the obtained samples were characterized by X-ray diffractometer (XRD) in the range of 10°–80° with Cu-Kα radiation (Philips X'pert Pro, λ =1.5406 Å). The morphology of the as-synthesized materials was identified through field emission scanning electron microscopy (FESEM, Gemini SEM 300) and transmission electron microscopy (TEM, Tecnai G2 F30). The elements of the representative surface area were further investigated by the element mapping technique. The X-ray photoelectron spectroscopy (XPS) data were evaluated on a Kratos AXIS Ultra DLD-600W XPS system with a monochromatic Al Ka (1486.6 eV) X-ray source. The specific surface area and pore size distribution were analyzed by Brunauer-Emmett-Teller instrument (BET, Quantachrome, autosorb iQ).
For preparation of the working electrode, the catalyst (5.0 mg), conductive acetylene black (1.0 mg), and Nafion solution (5.0 wt%, 30 μL) were dissolved in a 1 mL mixture solution of equimolar water and absolute ethanol under sonication. Afterwards, 10 μL catalyst inks were pipetted onto the glassy carbon (GC) electrode. The GC ring-disk electrode loaded with as-prepared catalyst materials served as the working electrode. The electrochemical potential was tested using Ag/AgCl as the reference electrode and carbon rod as the counter electrode. All electrolysis experiments were carried out in a standard three-electrode configuration connected to an electrochemistry workstation (Chenhua Instruments, CHI 760E) and a rotating-disk device (PINE, AFCPRBE) with the rotation rate of 1600 rpm.
In addition, all electrochemical measurements involved in the urea electro oxidation reaction are examined in 1.0 mol/L KOH +0.33 mol/L urea aqueous electrolyte unless otherwise noted. All linear sweep voltammograms are recorded at a scan rate of 5 mV/s with 95% iR correction to compensate the losses of ohmic potential drop resulted from the electrolyte resistance. Tafel slope is determined by the following equation linearly fitted with the overpotential and log (J): η = blog(J) + a, where η is the overpotential, b is the Tafel slope, and J refers to the current density. Electrochemical impedance spectroscopy (EIS) is carried out at a frequency between 0.1 Hz and 100 kHz. The effective electrochemical surface area (ECSA) is measured with the potential window ranging from 0.06 V to 0.16 V (vs. RHE) and the scan rate of 10~50 mV/s with the interval of 10 mV/s. Chronopotentiometry is conducted with a constant current density of 10 mA/cm2 for 23 h to assess the durability of catalyst.
The typical synthesis route of Ni2P/C-YS and the catalytic process towards UOR is schematically illustrated in Scheme 1. Firstly, the york-shell Ni-gly precursor is well designed and fabricated via a one-pot template-free solvothermal method [31-33]. The formation mechanism of york-shell nanostructures is mainly due to the Ostwald ripening process [34]. Specifically, the solid nanospheres with rough surfaces are obtained after the Ni ions integrate with glycerol molecules. During the next stage of the solvothermal reaction, the inner cores are gradually separated from the nanospherical shell, and continue to shrunk. With the extended reaction time, the voids between the inner cores and outer shells are formed, leading to the formation of the hollow york-shell architecture. Secondly, the Ni-gly precursor is transferred into the Ni2P/C-YS after phosphidation treatment at a high temperature (see the experiment sections), still maintaining the york-shell nanosphere architecture.
The crystallographic structures of the samples were unveiled by X-ray diffraction (XRD). As illustrated in Fig. 1a, a series of characteristic diffraction peaks at 40.6°, 44.6°, 47.3°, 54.2°, 54.8°, 66.2°, 72.7° and 74.8° in the XRD patterns of Ni2P/C-YS-300 and Ni2P/C-YS-350 are distinctly observed, indexing to (111), (201), (210), (300), (211), (202), (311) and (212) planes of the hexagonal Ni2P (JCPDS card No. 74-1385). Besides, the weak and broad peaks at 25.7° can be attributed to (002) crystal plane of graphite carbon (JCPDS card No. 41-1487), which confirms that the as-prepared samples are composed of Ni2P and carbon [28, 35]. In comparison, no obvious diffraction peak at 25.7° is detected both in the XRD patterns of Ni2P/C-YS-400 and Ni2P/C-YS-450, possibly due to the reason that the carbon elements of glycerate are transferred to the hydrocarbons after volatilization under the elevated temperature condition, resulting in a lower carbon content. As is well known, the higher annealing temperature favors the formation of the rich-P phase accompanied with the phase transition from Ni2P to Ni5P4, NiP or even NiP2 [36, 37]. Hence, it is understandable that Ni2P/C-YS-450 possesses the mixed phases, including hexagonal Ni2P phase and cubic NiP2 phase with the discernible diffraction peaks at 33.5° (200), 36.6° (210) and orthorhombic NiP phase with diffraction peaks at 50.9° (311). Noteworthy, a strong diffraction peak at 10.1° is discerned in sample Ni2P/C-YS-250 (Fig. S1 in Supporting information), being associated to the metal alkoxides of the Ni-gly precursor. Moreover, several unknown diffraction peaks suggest that 250 ℃ is not enough high for the formation of any phosphide [38]. Next, the Raman spectrum analysis was carried out to examine the formed carbon layers in Ni2P/C-YS-350. As shown in Fig. 1b, two different peaks at 1334 cm−1 and 1584 cm−1 are discerned, corresponding to the characteristic D-band and G-band of the carbonaceous material [39, 40]. The D-band (disordered phonon mode) is attributed to structural defects in the graphite plane, and the G-band (graphite band) is related to the E2g vibration mode of sp2 bond in graphitic carbon [27, 39]. The peak intensity ratio of the D-band to the G-band (ID/IG) is usually used to identify the disorder degree of the graphite structures. Since the calculated ID/IG ratio is 1.005, it signifies that the carbon layers in Ni2P/C-YS-350 have high quality, possessing high conductivity and facilitating the electron transfer during the electrochemical reaction [27, 41].
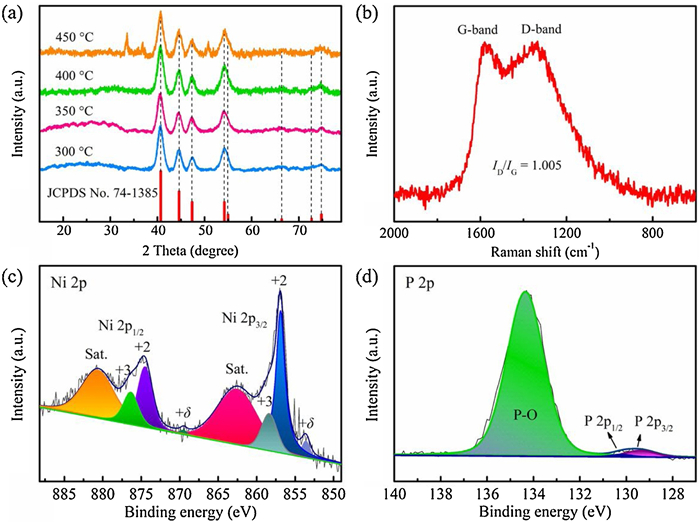
The surface chemical composition and electronic states of Ni2P/C-YS-350 were studied by X-ray photoelectron spectroscopy (XPS). Fig. S2a (Supporting information) illustrates the survey scan spectrum of Ni2P/C-YS-350, implying the coexistence of Ni, P, C and O. As shown in Fig. 1c, the high resolution Ni 2p spectrum of Ni2P/C-YS-350 is deconvoluted into six peaks. The binding energies of 856.9 eV and 874.5 eV with the satellite peaks (862.5 eV and 880.5 eV, marked as "Sat.") correspond to the Ni 2p3/2 and Ni 2p1/2, respectively, and being characteristic of the oxidation state of Ni2+ species [42, 43]. The signal peaks at 858.3 eV and 876.3 eV are due to the presence of the high-valence Ni. The faint peaks at 853.5 eV and 869.5 eV are ascribed to Niδ+ in Ni-P species, and the value of δ is assumed to be zero [20, 44]. As shown inFig. 1d, the P 2p spectrum of Ni2P/C-YS-350 exhibits three peaks, in which the identified peaks of P 2p3/2 (129.6 eV) and P 2p1/2 (130.3 eV) evidence the successful formation of phosphide-metal bonds [45, 46]. The sharp peak at 134.1 eV can be attributed to the oxidized P species on the surface [45, 46]. As shown in the high resolution spectrum of C 1s in Fig. S2b (Supporting information), three different peaks at 284.7, 285.7 and 288.7 eV are deconvoluted, which are generally attributed to the non-oxygenate carbon, carbon in C-O and carboxylate carbon, respectively, also confirming the presence of the coated carbon element [39, 47]. In addition, the X-ray photoelectron spectra for Ni-gly precursor were also identified and illustrated in Fig. S3 (Supporting information).
Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) were employed to observe the microscopic structural morphology of the as-prepared catalysts. Figs. S4a–d (Supporting information) display the low magnified FESEM images of Ni-gly precursor, indicating that the Ni-gly nanospheres with rough surface are well dispersed. Besides, some holes are identified in broken nanospheres in the high magnification image (Fig. S4c), indicating the hollow structure feature. Fortunately, a serial of the hollow yolk-shell nanospherical structures can be clearly discerned under 15 kV voltages (Fig. S4d). Fig. 2a shows the panoramic FESEM image of Ni2P/C-YS-350, which illustrates that the average size of the yolk-shell sphere is approximately 500 nm, clarifying that the next product of Ni-gly precursor successfully inherits their yolk-shell morphology. It is worthy mentioned that the as-prepared Ni2P/C-YS-350 consists of the inner spherical core and the surrounded spherical shell in the outer area, which can be clearly observed in the captured FESEM image of a broken nanosphere with a high magnification (Fig. 2b) and at 15 kV voltages (Fig. 2c). In comparison, the morphology of the control samples (i.e., the samples underwent different phosphidation treatment temperatures) were also disclosed, in which no significant change in diameter and surface roughness is identified (Figs. S5–S8 in Supporting information).

Fig. 2d represents the high-resolution transmission electron microscopy (HRTEM) image of Ni2P/C-YS-350, in which well-resolved lattice fringes with interplanar distances of 0.192 nm and 0.202 nm are discerned, corresponding to the (210) and (201) planes of Ni2P. The observed diffractive bright rings in the selected area electron diffraction (SAED) pattern (Fig. 2e) correspond to the (300), (210), (201) and (111) planes of the hexagonal Ni2P, being consistent with the XRD results and again confirming the polycrystalline nature of Ni2P. The elemental line scanning was conducted for the sample of Ni2P/C-YS-350. As shown in Fig. 2f, it shows that the spatial distribution of Ni, P and C elements uniformly distributes over the whole nanosphere. Of Note, the elemental signal of Ni in the inner core area is much stronger than that in the outer area, which further confirms the formation of the yolk-shell structure. Moreover, the energy-dispersive X-ray spectroscopy (EDS) further validates the fact that the introduction of P and C elements by converting Ni-gly precursor into Ni2P/C hybrid via the phosophidation process (Fig. S9 in Supporting information). The observed O element may be derived from glycerol reagent and the surface oxides product of the sample in the air. In addition, the pore structure and pore size distribution of Ni2P/C-YS-350 were characterized by the nitrogen adsorption/desorption isotherms from Brunauer-Emmett-Teller (BET) measurement. As shown in Fig. S10a (Supporting information), the specific surface area is calculated to be 3.6663 m2/g. Fig. S10b (Supporting information) illustrates that the pore size distribution is in the range of 2−18 nm and the average pore size of Ni2P/C-YS-350 derived from the desorption branch by Barrett-Joyner-Halenda (BJH) method is 12 nm, indicating their mesoporous feature, which can facilitate the transport of electrons and ions to promote the improvement of catalytic performance.
The catalytic performance towards UOR was implemented on a rotating disk electrode in the standard three-electrode system in the electrolyte of 1 mol/L KOH and 0.33 mol/L urea. As shown in Fig. 3a, the linear sweep voltammetry (LSV) curves with 95% iR compensation reveal that Ni2P/C-YS-350 exhibits the lowest potential (1.354 V) at a current density of 10 mA/cm2, which is lower than the control samples prepared with different phosphidation temperatures (i.e., 1.373 V for Ni2P/C-YS-250, 1.356 V for Ni2P/C-YS-300, 1.360 V for Ni2P/C-YS-400, 1.363 V for Ni2P/C-YS-450). Moreover, the potentials of Ni2P/C-YS at the current density of 50 mA/cm2 are recorded as 1.444, 1.374, 1.366, 1.382 and 1.85 V at the phosphidation temperature ranging from 250 ℃ to 450 ℃. The above advanced catalytic activity can be explained by the great difference in crystalline phase of as-prepared samples under different phosphidation temperature. The crystalline phase impurities (e.g., the various kinds of nickel phosphide phases at 450 ℃ and the traces of large numbers of Ni-gly precursor at 250 ℃) significantly decrease the trivalent Ni species, leading to the deteriorated catalytic performance. Besides, due to the synergistic effect between active Ni2P crystalline phase and doped carbon particles, Ni2P/C-YS-300 and Ni2P/C-YS-350 possess good electron conductivity, facilitating the electronic transport and thereby achieving the promoted catalytic activity toward UOR. In order to further evaluate the catalytic performance of Ni2P/C-YS-350, the electrocatalytic performance of the commercial Pt/C electrode and the corresponding Ni-gly precursor were also examined and illustrated in Fig. S11a (Supporting information). The UOR activities of Ni2P/C-YS-350 show the lowest potential and highest current density among the above testing materials. For example, the potentials of Ni-gly precursor and Pt/C are 1.426 V and 1.742 V respectively, when the current density is 50 mA/cm2. As shown in Table S1 (Supporting information), the comparison results reveal that our Ni2P/C-YS-350 still exhibits the excellent UOR performance among the recently reported electrocatalysts. In addition, to investigate the UOR kinetics of the Ni2P/C-YS-350, Tafel slopes analysis was carried out to investigate the UOR kinetics of the as-prepared samples. As show in Fig. 3b, Ni2P/C-YS-350 exhibits the lowest Tafel slope of 45.3 mV/dec, remarkably lower than those of the control samples with different phosphidation temperatures (e.g., 53.4 mV/dec for Ni2P-YS-300, 60.9 mV/dec for Ni2P-YS-400, 75.0 mV/dec for Ni2P-YS-450, 176.2 mV/dec for Ni2P-YS-250), also implying the enhanced UOR activity of Ni2P/C-YS-350 by the virtue of the active Ni2P crystallinity and appropriate carbon contents. Compared with Pt/C and Ni-gly precursor, the Tafel slope of Ni2P/C-YS-350 is also much lower than those of f Ni-gly precursor (193.0 mV/dec) and Pt/C (235.9 mV/dec), indicating the fast catalytic reaction kinetics (Fig. S11b in Supporting information).

The electrochemical impedance spectroscopy (EIS) was an important pathway to evaluate the charge transfer behaviors of electrochemical catalysts. The Nyquist plots from the EIS fitting with the similar Randles equivalent circuit are shown in Fig. 3c. As is well known, the semicircle in the high-frequency range ascribed to charge transfer resistance (Rct) at the electrode/electrolyte interface [48]. Among the as-prepared samples, the Rct value decreases as the following order: Ni2P/C-YS-350 < Ni2P/C-YS-300 < Ni2P/C-YS-400 < N2P/C-YS-450 < Ni2P/C-YS-250. The smallest Rct of Ni2P/C-YS-350 reveals the smaller charge transfer resistance and the better charge transport kinetics in Ni2P/C-YS-350 catalyst. Particularly, the Nyquist plots in the low frequency range for Ni2P/C-YS-350 and Ni-gly are also collected. Fig. S11c (Supporting information) displays the larger semicircle diameter of Ni-gly (37 O) than that of Ni2P/C-YS-350. In addition, the UOR catalytic activity was further examined through the effective electrochemical surface area (ECSA). All the electrochemical double layer capacitance (Cdl) can be determined from the CV measurements at different scan rates in nonfaradaic potential region in Fig. S12 (Supporting information). In Fig. 3d, it exhibits that Ni2P/C-YS-350 exhibits the largest Cdl value of 0.48 μF/cm2 among the control samples (e.g., 0.17 μF/cm2 for Ni2P/C-YS-250, 0.34 μF/cm2 for Ni2P/C-YS-300, 0.29 μF/cm2 for Ni2P/C-YS-400, 0.20 μF/cm2 for N2P/C-YS-450). On the other hand, since the Cdl value of Ni-gly precursor is 0.37 μF/cm2, and lower than that of Ni2P/C-YS-350 (Fig. S11d in Supporting information), it evidences the increasing number of effective active sites upon the phosphidation treatment of Ni-gly precursor towards forming Ni2P/C-YS-350. Apart from the UOR catalytic activity, the long-term stability also needed to be seriously concerned, especially from practical application perspective. As shown in Fig. 3e, the durability test is carried out by means of the chronoamperometry measurements (j-t) at a constant current density of 50 mA/cm2. The slightly increase in the potential of Ni2P/C-YS-350 is identified during the UOR electrocatalysis process after 23 h, indicating that Ni2P/C-YS-350 shows the superior catalytic UOR stability.
In order to investigate the effect of urea concentration on the UOR process, the electrochemical performance test was performed on Ni2P/C-YS-350 in the alkaline electrolyte with different urea concentrations. Fig. 3f shows the LSV curves of Ni2P/C-YS-350 measured in the electrolytes containing pure 1 mol/L KOH, 1 mol/L KOH and 0.1 mol/L urea, 1 mol/L KOH and 0.33 mol/L urea, 1 mol/L KOH and 0.5 mol/L urea. It can be found that the oxygen evolution reaction occurs at the potential of 1.546 V at 10 mA/cm2 when no urea is involved. The current density recorded in 0.1 mol/L urea is 151.5 mA/cm2; interestingly, it significantly increases to 252.5 mA/cm2 with the existence of 0.33 mol/L urea at 1.45 V (vs. RHE) under natural diffusion. Nonetheless, it should be noted that the current density decreases to 206.0 mA/cm2 when the urea is further increased to 0.5 mol/L urea. In light of these facts, the gradient effect of the urea concentration can be highlighted toward the UOR performance, implying that Ni2P/C-YS-350 exhibits the highest performance towards urea oxidation in electrolyte solution mixture of 1 mol/L KOH and 0.33 mol/L urea.
The in-depth mechanistic analysis toward UOR electrocatalytic process has been widely studied. It is well acknowledged that the UOR process for the nickel-based catalysts involves successive electrochemical-chemical (EC) mechanism via a six-electron transfer reaction pathway. During the electrochemical process, Ni2+ can be easily passivated to form Ni(OH)2 in the strong alkaline solution, and further oxidized into Ni3+ (e.g., formation of NiOOH). NiOOH, working as the essential active species, subsequently reacts with urea molecules to generate N2 and CO2 via several chemical steps, simultaneously leading to the decrease of the valence state of Ni. To gain further insight into the real active sites and electro-oxidation process for UOR, the Ni2P/C-YS sample after UOR tests was examined with the XPS measurement (Fig. S13 in Supporting information) and carefully compared with the XPS results before UOR test. As shown in Fig. S13b, the divalent Ni peaks (855.9 and 872.9 eV) are weaken and downshifted compared to the pristine one; meanwhile, the peaks of Ni0 also are not detected any more. In case of the high-valence Ni, the peaks (856.6 and 873.9 eV) become much stronger, confirming the fact that low-valence Ni species are inevitably oxidized to the trivalent Ni species (NiOOH) during the UOR process [27, 35]. As is stated aforementioned, the resultant NiOOH should be the main catalytic species toward UOR. Fig. S13c depicts the core-level P 2p spectrum, in which it is found that nearly all the peaks disappear after the UOR test, suggesting the conversion of phosphorus into phosphate during the UOR reaction. The accompanied dissolution of P element can be attributed to the in-situ superficial oxidative transformation, giving rise to the formation of nickel oxyhydroxide layers during the UOR activation process [27, 35].
As is well known, the essence of the electrocatalytic reactions can be regarded as the coupling interactions between the catalyst surface and the adsorbates, ultimately giving rise to the formation of the fully-filled bonding states and the empty or partially-filled anti-bonding states of the electronic structures [49]. In previous studies, the adsorption energy of urea molecules can be optimized via the metal elements doping or oxygen vacancies in modulating the electronic structures of Ni sites according to the experimental results and DFT calculations [16, 50-52]. In our work, the elevated UOR performance is benefited from the synergy between Ni2P and C with the particular architecture. At beginning, the OH− tends to break the NH2 groups of urea molecules upon the approach of urea on the surface of Ni2P/C-YS sample under the certain potential, leading to the transport of the released electrons to NiOOH and the formation of H2O. In the next step, NH2 group converts into the N species adsorbed on the exposed Ni sites after breaking the C-N bond of urea molecule, giving rise to nitrogen and carbon dioxide gas, and the highly active NiOOH return back to Ni2+. Owing to the carbon additive serving as the electron acceptor, the electrons can be transported from Ni site to C site, enabling Ni be in the high-valence state for further catalytic oxidation. Meanwhile, the π → π* transition of the C=C bond in sp2 domain occurs after the electron captured by C sites, which can be evidenced from the newly appeared peak at 291.7 eV shown in C 1s spectrum in Fig. S13d [53]. In addition, benefiting from the yolk-shell structure, the Ni2P/C has numerous exposed active sites and possesses unique electronic structures, endowing the high conductivity for fast electron transfer along the surface of catalysts and lowering the carbon dioxide adsorption/desorption barrier for fast reaction kinetics, ultimately boosting the UOR process.
In conclusion, we have prepared a series of Ni2P/C-YS composites with the regulated phosphidation temperatures. The boosted UOR activity is benefited from the unique hollow yolk-shell configuration, in which the exposed numerous interfaces and large electrochemical active surface area allow the electron transfers between Ni2P and coated carbon nanoparticles. By the virtue of more conductive electrode materials upon the introduction of carbon additives, the UOR catalytic performance exhibit low potential of 1.366 V at a current density of 50 mA/cm2, small Tafel slope of 45.3 mV/dec and a negligible potential decay in durability test. The UOR process catalyzed by Ni2P/C-YS composites experience a successive electrochemical-chemical reaction mechanism, in which urea oxidation occurs accompanying with the redox of NiOOH at high potentials. The present work provides a profound perspective for the rational design and preparation of nonprecious metals/carbon hybrid materials with the tuned hollow yolk-shell morphology in the further areas of overall water splitting with urea.
The authors report no declarations of interest.
This work is financially supported by the National Key Research and Development Program of China (No. 2017YFE0120500), the National Natural Science Foundation of China (Nos. 51804223 and 51972129), CAS Key Laboratory of Nano-Bio Interface (No. 19ZY01), the South Xinjiang Innovation and Development Program of Key Industries of Xinjiang Production and Construction Corps (No. 2020DB002), the Scientific Research Foundation of Wuhan Institute of Technology (No. K201761) the Fundamental Research Funds for the Central Universities (Nos. HUST 2018KFYYXJJ051, 2019KFYXMBZ076), C. Wang acknowledges the Hubei "Chu-Tian Young Scholar" Program.
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.040.
N. Senthilkumar, G. Gnana kumar, A. Manthiram, Adv. Energy Mater. 8(2018) 1702207. doi: 10.1002/aenm.201702207
Y.X. Wang, C.F. Liu, M.L. Yang, et al., Chin. Chem. Lett. 28(2017) 60-64. doi: 10.1016/j.cclet.2016.05.025
J. Maya-Cornejo, R. Carrera-Cerritos, D. Sebastián, et al., Int. J. Hydrogen Energy 42(2017) 27919-27928. doi: 10.1016/j.ijhydene.2017.07.226
G. Wang, Z. Wen, Nanoscale 10(2018) 21087-21095. doi: 10.1039/C8NR06740F
W. Yang, X. Yang, B. Li, et al., J. Mater. Chem. A 7(2019) 26364-26370. doi: 10.1039/C9TA06887B
N. Kakati, J. Maiti, K.S. Lee, et al., Electrochim. Acta 240(2017) 175-185. doi: 10.1016/j.electacta.2017.04.055
K. Ye, G. Wang, D. Cao, et al., Top. Curr. Chem. 376(2018) 42. doi: 10.1007/s41061-018-0219-y
A.V. Munde, B.B. Mulik, P.P. Chavan, et al., Electrochim. Acta 349(2020) 136386. doi: 10.1016/j.electacta.2020.136386
C. Xiao, S. Li, X. Zhang, et al., J. Mater. Chem. A 5(2017) 7825-7832. doi: 10.1039/C7TA00980A
Z. Zhao, J. Zhao, H. Wang, et al., Int. J. Hydrogen Energy 45(2020) 14199-14207. doi: 10.1016/j.ijhydene.2019.11.007
X. Zhu, X. Dou, J. Dai, et al., Angew. Chem. Int. Ed. 55(2016) 12465-12469. doi: 10.1002/anie.201606313
Y. Tong, P. Chen, M. Zhang, et al., ACS Catal. 8(2018) 1-7. doi: 10.1021/acscatal.7b03177
G. Ma, Q. Xue, J. Zhu, et al., Appl. Catal. B: Environ. 265(2020) 118567. doi: 10.1016/j.apcatb.2019.118567
W. Simka, J. Piotrowski, G. Nawrat, Electrochim. Acta 52(2007) 5696-5703. doi: 10.1016/j.electacta.2006.12.017
J. Yoon, D. Lee, Y.N. Lee, et al., J. Power Sources 431(2019) 259-264. doi: 10.1016/j.jpowsour.2019.05.059
X. Yan, W.D. Zhang, Q.T. Hu, et al., Int. J. Hydrogen Energy 44(2019) 27664-27670. doi: 10.1016/j.ijhydene.2019.09.004
D. Yang, L. Yang, L. Zhong, et al., Electrochim. Acta 295(2019) 524-531. doi: 10.1016/j.electacta.2018.10.190
P. Xiong, X. Ao, J. Chen, et al., Electrochim. Acta 297(2019) 833-841. doi: 10.1016/j.electacta.2018.12.043
R.P. Forslund, J.T. Mefford, W.G. Hardin, et al., ACS Catal. 6(2016) 5044-5051. doi: 10.1021/acscatal.6b00487
C. Teng, N. Zhang, X. Gao, et al., Mater. Today Energy 11(2019) 192-198. doi: 10.1016/j.mtener.2018.11.008
Q. Wang, Z. Liu, H. Zhao, et al., J. Mater. Chem. A 6(2018) 18720-18727. doi: 10.1039/C8TA06491A
Y. Jin, C. Zhao, L. Wang, et al., Int. J. Hydrogen Energy 43(2018) 3697-3704. doi: 10.1016/j.ijhydene.2018.01.008
Y. Zhang, L. Sun, Y. Li, et al., J. Electroanal. Chem. 873(2020) 114441. doi: 10.1016/j.jelechem.2020.114441
M. Peroni, G. Mancino, E. Baráth, et al., Appl. Catal. B: Environ. 180(2016) 301-311. doi: 10.1016/j.apcatb.2015.06.042
P.M. de Souza, C.V.M. Inocêncio, V.I. Perez, et al., Catal. Today 356(2020) 366-375. doi: 10.1016/j.cattod.2019.08.028
B. Konkena, J. Masa, A.J.R. Botz, et al., ACS Catal. 7(2017) 229-237. doi: 10.1021/acscatal.6b02203
M. Wang, M. Lin, J. Li, et al., Chem. Commun. 53(2017) 8372-8375. doi: 10.1039/C7CC03558F
D. Yang, Y. Gu, X. Yu, et al., ChemElectroChem 5(2018) 659-664. doi: 10.1002/celc.201701304
Q. Gan, X. Cheng, J. Chen, et al., Electrochim. Acta 301(2019) 47-54. doi: 10.1016/j.electacta.2019.01.150
X. Ji, Y. Zhang, Z. Ma, et al., ChemSusChem 13(2020) 5004-5014. doi: 10.1002/cssc.202001185
G.D. Park, J.H. Hong, J.K. Lee, et al., Nanoscale 11(2019) 631-638. doi: 10.1039/C8NR08638A
C. Zhang, H. Yang, D. Zhong, et al., J. Mater. Chem. A 8(2020) 9536-9544. doi: 10.1039/D0TA00962H
J. Li, J. Li, D. Yan, et al., J. Mater. Chem. A 6(2018) 6595-6605. doi: 10.1039/C8TA00557E
L. Wang, Z. Han, Q. Zhao, et al., J. Mater. Chem. A 8(2020) 8612-8619. doi: 10.1039/D0TA02568B
G. Li, J. Wang, J. Yu, et al., Appl. Catal. B: Environ. 261(2020) 118147. doi: 10.1016/j.apcatb.2019.118147
Y. Yan, J. Lin, K. Bao, et al., J. Colloid Interface Sci. 552(2019) 332-336. doi: 10.1016/j.jcis.2019.05.064
D. Li, K. Senevirathne, L. Aquilina, et al., Inorg. Chem. 54(2015) 7968-7975. doi: 10.1021/acs.inorgchem.5b01125
L. Shen, L. Yu, H.B. Wu, et al., Nat. Commun. 6(2015) 6694. doi: 10.1038/ncomms7694
B. Zhang, Y.H. Lui, A.P.S. Gaur, et al., ACS Appl. Mater. Interfaces 10(2018) 8739-8748. doi: 10.1021/acsami.8b00069
J. Yu, Q. Li, N. Chen, et al., ACS Appl. Mater. Interfaces 8(2016) 27850-27858. doi: 10.1021/acsami.6b10552
J.Y. Wang, T. Ouyang, N. Li, et al., Sci. Bull. 63(2018) 1130-1140. doi: 10.1016/j.scib.2018.07.008
C. Yu, F. Xu, L. Luo, et al., Electrochim. Acta 317(2019) 191-198. doi: 10.1016/j.electacta.2019.05.150
P. Jiang, Q. Liu, X. Sun, Nanoscale 6(2014) 13440-13445. doi: 10.1039/C4NR04866K
M. Qian, S. Cui, D. Jiang, et al., Adv. Mater. 29(2017) 1704075. doi: 10.1002/adma.201704075
Y. Zhang, X. Gao, L. Lv, et al., Electrochim. Acta 341(2020) 136029. doi: 10.1016/j.electacta.2020.136029
Y. Zhang, J. Xu, Y. Ding, et al., Int. J. Hydrogen Energy 45(2020) 17388-17397. doi: 10.1016/j.ijhydene.2020.04.213
Y. Zhang, C. Wang, Mater. Today Energy 16(2020) 100406. doi: 10.1016/j.mtener.2020.100406
Y. Zhang, J. Xu, L. Lv, et al., Nanoscale 12(2020) 10196-10204. doi: 10.1039/D0NR01809K
Z. Chen, Y. Song, J. Cai, et al., Angew. Chem. Int. Ed. 57(2018) 5076-5080. doi: 10.1002/anie.201801834
M.S. Wu, Y.J. Sie, S.B. Yang, Electrochim. Acta 304(2019) 131-137. doi: 10.1016/j.electacta.2019.02.100
J. Xie, W. Liu, F. Lei, et al., Chem. : Eur. J. 24(2018) 18408-18412. doi: 10.1002/chem.201803718
C. Wang, H. Lu, Z. Mao, et al., Adv. Funct. Mater. 30(2020) 2000556. doi: 10.1002/adfm.202000556
B.C. Mallick, C.T. Hsieh, K.M. Yin, et al., Nanoscale 11(2019) 7833-7838. doi: 10.1039/C8NR10118C

Scheme 1 Schematic diagram of the preparation of a yolk-shell nanostructural catalyst of Ni2P/C.

Figure 1 (a) XRD patterns of Ni2P/C-YS. (b) Raman spectrum of Ni2P/C-YS-350. (c, d) XPS spectra of Ni2P/C-YS-350 for Ni 2p and P 2p, respectively.

Figure 2 (a, b) low magnification and high magnification FESEM images of Ni2P/C-YS-350. (c) FESEM image of Ni2P/C-YS-350 at 15kV voltages. (d, e) HRTEM image and SAED pattern of Ni2P/C-YS-350. (f) HAADF-STEM image of Ni2P/C-YS-350 and the corresponding EDS elemental mapping image.

Figure 3 (a, b) LSV curves with iR compensation and Tafel slope curves of Ni2P/C-YS. (c) Plots of the current density (at 1.133 V) vs. the scan rate. (d) Nyquist plots of Ni2P/C-YS. (e) Chronopotentiometric measurements of Ni2P/C-YS-350 at 10 mA/cm2. (f) LSV curve of Ni2P/C-YS-350 under different concentrations of urea.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: