引用本文:
段秉亚, 王天元, 孙应飞. 计算机辅助的金属蛋白设计与改造研究进展[J]. 化学通报,
2019, 82(3): 221-230.

Citation: Duan Bingya, Wang Tianyuan, Sun Yingfei. Research Progress in Computer Aided Metalloprotein Design and Engineering[J]. Chemistry, 2019, 82(3): 221-230.
Citation: Duan Bingya, Wang Tianyuan, Sun Yingfei. Research Progress in Computer Aided Metalloprotein Design and Engineering[J]. Chemistry, 2019, 82(3): 221-230.
计算机辅助的金属蛋白设计与改造研究进展
English
Research Progress in Computer Aided Metalloprotein Design and Engineering
Abstract:
Metalloproteins are proteins containing metal ions, they play important roles in biology system due to their special catalytic activity. A lot of study has been done to elucidate their structural and functional mechanism. Based on the active site mimic of existing metalloprotein, artificially designed metalloproteins with specific structure and function can be achieved through computer aided protein design method. In this paper, research progress in computer aided metalloprotein design and engineering was reviewed. The general principle for introducing metal ion binding site and protein activity enhancement was summarized. We also presented current problems and challenges in this field, along with future directions and probable breakthrough point.
-
金属蛋白是含有金属离子的蛋白的统称,在不同的金属蛋白中,金属离子(如铁、铜、锌、钴、锰等)以不同的价态、配位形式和辅因子形式存在。金属离子独特的化学性质赋予了金属蛋白众多特殊的催化活性,在生物体和生物地球化学循环中发挥着重要作用。例如,Cu/Zn超氧化物歧化酶(SOD)含有Cu/Zn离子,在人体中广泛存在,其具有的氧化还原性质可以清除体内活性氧自由基(ROS),防止细胞的氧化损伤,维持体内免疫系统的氧化还原平衡[1];RuBisco蛋白[2]是世界上丰度最高的蛋白质,在光合作用中催化了固碳暗反应的第一步,蛋白中的Mg2+固定了底物、二氧化碳及关键的甲酸赖氨酸翻译后修饰残基,对于催化二氧化碳固定的活性至关重要;细胞色素P450(CYP)蛋白[3]含有金属离子-血红素辅因子,在人体代谢中发挥重要作用,许多药物分子进入人体后在细胞色素P450的催化下发生反应,影响着药物分子的代谢、毒性、药效等性质,是药物化学家的重要研究对象;在生物地球化学循环中,一些阴离子(如亚硝酸根、亚硫酸根等)的还原酶[4]含有血红素和[4Fe-4S]簇立方体,可以催化多电子还原反应,将高价态的氮、硫等元素还原为低价态,对于维持元素地球化学循环和生态环境稳定具有重要意义。
鉴于金属蛋白具有独特的催化活性,利用人工蛋白模拟金属蛋白的活性是化学生物学家一直以来追寻的目标。这项研究可以检验、加深人们对天然金属蛋白结构-功能关系的理解,同时设计得到的各种活性的金属蛋白可以应用在生物学研究甚至工业生产的各个方面,具有理论和现实的双重意义。计算机辅助设计技术结合了计算化学和计算生物学的方法,大大提高了人工金属蛋白设计的成功率,加速了此领域的研究进展。
1. 计算机辅助蛋白设计
1.1 计算机辅助蛋白设计的定义
蛋白设计是指通过理性设计获得特定的氨基酸序列,以构建指定的蛋白结构或实现催化功能的方法。一般认为蛋白的功能是由其结构决定的,而结构是由一级氨基酸序列决定的。不同的氨基酸序列导致蛋白折叠成不同的三维结构,具有不同的功能。与此同时,疏水相互作用是蛋白折叠的驱动力,因为疏水残基暴露在溶剂中是能量不利的状态,疏水残基在蛋白内部互相堆积可以有效减少与外部溶剂的接触,是一种低能量的有利状态。疏水残基的数量、种类、分布位置等都会影响蛋白折叠后的三维结构。天然氨基酸(NAA)有20种,因此一个含有100个氨基酸的小蛋白可能的氨基酸组合有20100≈1.3×10130种,要从这样一个天文数字的组合中找出目标序列是一项极其艰巨的任务。蛋白设计可以分为两类:从头设计(de novo design)和对已有蛋白的改造再设计。在从头设计的任务中,科学家需要获得的目标蛋白不同于已知的任何蛋白序列,但具有指定的结构或功能;在对已有蛋白的改造中,一般是改造底物结合位点的序列,获得特定的蛋白活性或者改变底物特异性。
随着计算机技术的发展、计算能力的提高以及先进算法的出现,近年来在利用计算机辅助进行蛋白设计领域取得了很多重要进展。一方面分子动力学模拟、量子化学计算、QM/MM等技术的发展可以让人们以更加准确的方式计算蛋白折叠的自由能面、蛋白-配体之间的相互作用以及催化过渡态的能量变化[5];另一方面高性能计算集群、分布式计算、GPU加速、云计算等技术的应用赋予了空前强大的计算能力,使得大规模进行蛋白设计的计算模拟成为了可能[6];第三,得益于许多先进算法的涌现,科学家可以更快地确定合适的蛋白骨架[7],更加准确地预测蛋白折叠后的构象[8],更为精准地预测底物在结合位点的作用模式[9],大大提高了蛋白设计的准确率。
1.2 用于蛋白设计的软件
早期的蛋白设计软件有InsightⅡ/Discovery(MSI)等,被用来进行螺旋束类型蛋白的设计[10],利用分子力场模型进行能量最小化。Dezymer软件被用来在蛋白骨架上引入不同氨基酸的旋转异构体(rotamer)以寻找特定的结合位点构型[11],这种算法可以用来在已知蛋白骨架上根据指定的构型寻找特定的金属离子结合位点。类似的Metal search软件被设计用来寻找具有四面体构型金属结合位点的蛋白[12]。Rosetta蛋白设计软件[13, 14]具有多种功能,其中之一为真正的从头设计蛋白,即根据已有的蛋白短片段(fragment)数据库通过蒙特卡洛方法组装寻找具有类似天然构型的蛋白,设计得到指定三维结构的蛋白;另一个功能为蛋白再设计,即根据催化反应的过渡态构型从已知的蛋白骨架数据库中寻找能容纳过渡态构型的蛋白,以实现底物特异性的改变或产生新的催化功能[15, 16]。
对于金属蛋白的设计,底物结合位点除了结合反应底物外,还有金属离子。为了设计得到具有特定活性的金属蛋白,首先需要考虑如何将金属离子引入蛋白:可以利用氨基酸残基(包括天然氨基酸NAA和非天然氨基酸UAA)直接与金属离子配位结合,也可以将结合了金属离子的辅因子引入蛋白。其次需要选择合适的蛋白骨架:要求蛋白骨架具有特定的空间构型以便在其中引入合适的金属离子结合位点,同时蛋白骨架需要有一定的稳定性[17],引入的突变不会对蛋白整体结构造成大的扰动或导致错误折叠。蛋白骨架可以从已有的蛋白结构数据库(如PDB)中搜寻,也可以通过从头设计的方法来获得具有特定拓扑结构的蛋白。第三,设计得到的金属蛋白往往活性较低,需要对其进行改造:一方面可以改造底物或辅因子结合位点的残基,增强结合力,另一方面通过改变与金属离子配位的氨基酸的性质或者改变金属离子与底物配位的方式可以获得新的活性。
2. 金属离子的引入——利用蛋白上的氨基酸残基直接与金属离子配位结合
2.1 在已有的蛋白骨架上构建金属离子结合口袋
2.1.1 利用天然氨基酸构建金属离子结合口袋
在金属蛋白中,不同的金属离子与蛋白的配位形式种类多样,即使同一种金属离子也有不同的配位方式,金属离子本身的性质和配位方式共同决定了金属蛋白的催化性质。与金属离子配位的氨基酸有组氨酸、天冬氨酸、谷氨酸、半胱氨酸等,设计者需要在蛋白合适的位点引入这些氨基酸以获得螯合金属离子的能力。
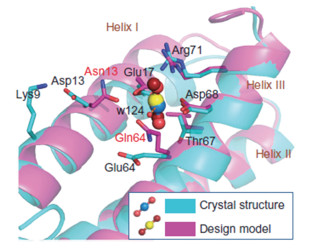
2014年,Zhou等[18]用计算机辅助设计得到了以fmol/L级别的Kd值特异性结合铀酰离子(UO2+)的蛋白,用于从海水中富集提取铀元素,其结构如图 1所示。他们首先从天然存在的铀酰离子配合物中提取了铀酰离子与氧原子配位的特征,从12173个蛋白骨架中构建氧原子和氢原子库,然后通过大规模计算筛选算法URANTEIN搜索符合配位条件的蛋白骨架。同时作者针对铀酰离子配位体系设计了特殊的力场和打分模型,最终根据计算结果进行筛选,成功得到了Kd值为37nmol/L的蛋白。进一步引入稳定蛋白结构的突变Leu67Thr和负电氨基酸突变Asn13Asp、Gln64Glu后得到了Kd值为7.4fmol/L的超级结合蛋白,表明带负电的基团对于铀酰离子的结合至关重要。该研究成功的关键在于高效准确地筛选特定配位构型蛋白骨架的算法以及针对具体问题开发的特定力场和打分模型。
图 1
2018年,Bozkurt等[19]利用遗传算法在只有56个氨基酸的小蛋白GB1上引入了四面体构型的锌离子结合位点,同时优化改造了其他位点以加强蛋白疏水核心的堆积作用,最终得到了具有高度热稳定性的蛋白,Tm值比野生型提高了36℃。作者使用了多轮计算评估的策略:第一轮计算中,评估了11个可能的组氨酸位点,考虑旋转异构体,共有2411种可能的情况,通过适应函数的得分最终确定了23、45和50为最好的组氨酸突变位点;第二轮计算中,固定23H和50H,通过遗传算法优化其他锌离子结合位点;第三轮计算中,优化疏水核心堆积位点,确定最终的序列。结果表明,这种多轮计算优化的策略可以确定合适的锌离子结合位点,同时可以提高蛋白的热稳定性。作为一个模型蛋白,GB1很适合用于金属蛋白改造的研究,早在1995年Klemba等[20]就通过计算设计得到了一个GB1蛋白的突变体,具有His3Cys-Zn(Ⅱ)结合位点,并且通过NMR测定并证实了设计的正确性。
2.1.2 利用非天然氨基酸构建金属离子结合口袋
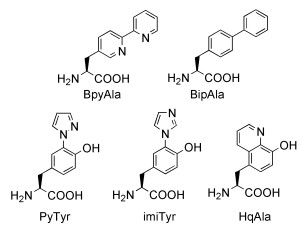
基因密码子扩展技术[21]允许科学家在目标蛋白的特定位点引入具有特殊反应活性的非天然氨基酸。通过在生物体内引入一套外源的tRNA/氨酰tRNA合成酶正交系统,可以实现基因编码特定的非天然氨基酸[21, 22]。目前已经有超过100种具有不同化学结构的非天然氨基酸通过基因编码的方式被插入到蛋白中。相比于自然界的20种天然氨基酸,非天然氨基酸具有特殊的化学反应活性或催化活性,为人工蛋白酶的设计提供了全新的思路和工具。例如,在最近的一项研究中,Roelfes等[23]利用对氨基苯丙氨酸特殊的催化活性设计得到了可以催化形成腙和肟的人工蛋白,显示了利用非天然氨基酸设计人工蛋白的巨大潜力。数个具有特殊金属螯合性质的非天然氨基酸及其类似物已经被基因编码到蛋白[24~27],如图式 1所示,为基于非天然氨基酸构建金属离子结合位点提供了很好的出发点。
图式 1
 图式 1. 具有金属离子螯合性质的非天然氨基酸及其类似物的结构式Scheme 1. Chemical structure of metal-chelating UAA and analogues
图式 1. 具有金属离子螯合性质的非天然氨基酸及其类似物的结构式Scheme 1. Chemical structure of metal-chelating UAA and analogues联吡啶丙氨酸(BpyAla)含有两个吡啶环,本身具有螯合金属离子的性质,为金属蛋白的设计提供了很好的出发点。早在2007年,Schultz等[24]通过基因密码子扩展技术实现了在目标蛋白中位点特异性地插入BpyAla。作为一项概念验证性的应用,他们[28]利用基因密码子扩展技术将BpyAla插入到大肠杆菌降解物激活蛋白CAP与DNA接触界面的K26位点,使CAP蛋白在铜离子存在时具有氧化切割DNA的活性。
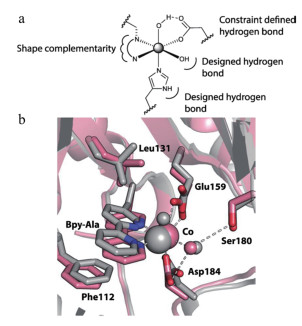
2013年Mills等[29]利用BpyAla结合金属离子的性质,通过Rosetta软件设计出了能结合二价金属离子的蛋白,利用基因密码子扩展技术表达获得了位点特异性插入BpyAla的设计蛋白,通过实验测得对锌离子的Kd值为40pmol/L。BpyAla本身含有两个吡啶环,两个氮原子的位置和取向利于结合金属离子,另外BpyAla本身为电中性,可以在蛋白内部的疏水结合位点引入金属离子,很好地避免了引入极性带电氨基酸造成的蛋白稳定性降低。他们首先定义了锌离子所有可能的配位形式,利用rosetta软件在指定蛋白骨架中进行搜索,通过两轮迭代设计获得了具有原子级别精确度的结构,如图 2所示。在第一轮设计中,晶体结构显示含有BpyAla的环(loop)向外侧翻转,偏离了预期设计的结构,这表明引入的氨基酸位点最好选择在较为稳定的二级结构区,如α-螺旋、β-折叠等,不会对蛋白结构造成较大的扰动。该工作证明了可以通过计算机辅助的方法设计得到原子级别精确度的金属离子结合位点,为实现高催化活性蛋白的设计打下了基础。此蛋白只能结合金属离子,没有催化活性,但为有活性金属蛋白的设计提供了很好的出发点。
图 2

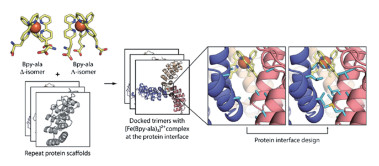
在上述工作中,作者观察到在蛋白上的BpyAla与溶液中的BpyAla螯合亚铁离子后形成了三聚物,说明[Fe(Bpy-ala)3]2+本身具有很高的稳定性。受此启发,Mills等[30]利用BpyAla螯合金属离子的性质,基于rosetta软件的算法设计了具有对称性的自组装同源三聚体蛋白,[Fe(Bpy-ala)3]2+复合物作为“桥梁”连接单体之间的接触界面。具体流程见图 3,首先从小分子晶体模型中提取了三个BpyAla螯合一个亚铁离子的配位构型,然后在具有三重对称性的重复蛋白骨架中利用对接算法确定可以容纳此配位构型的合适位点,同时保证接触界面的残基之间没有空间位阻。之后计算优化接触界面的残基,使得界面之间具有最高的互补程度。晶体结构显示设计得到的蛋白具有原子级别的准确度,为具有特殊光学或光物理性质的蛋白设计开辟了新的途径。作为另一项应用,他们将BpyAla插入到三聚螺旋束蛋白1ZN17的三聚物接触界面以结合亚铁离子,使得Tm值提高到超过95℃,是提高螺旋束蛋白热稳定性的有效方案[31]。
图 3

2015年,Pearson等[5]报道了通过rosetta软件计算设计得到的“蛋白瓶”,可以稳定维持BpyAla的类似物联二苯丙氨酸(BipAla)中两个苯环共平面的高能量状态,为稳定催化过程中高能量的过渡态提供了有价值的参考。
自然界中的金属蛋白具有众多独特的催化活性,人工设计金属酶模拟天然金属酶的活性具有重要意义。Roelfes课题组[32~35]以LmrR蛋白为研究对象,利用蛋白二聚体接触界面的平坦疏水空腔作为催化位点,引入金属离子和催化底物,设计得到具有多种催化活性的金属蛋白。2015年,他们[33]利用BpyAla结合铜离子的性质,在LmrR二聚体界面位点引入BpyAla得到了具有催化不对称Friedel-Crafts烷基化反应活性的铜蛋白。尝试在不同位点引入BpyAla并且改变底物结合位点周围的氨基酸,最终得到了ee值高达83%的突变体。各种突变体的测试结果表明第二配位层的氨基酸对于催化活性影响很大,π-堆积作用可能调整了底物结合方向。
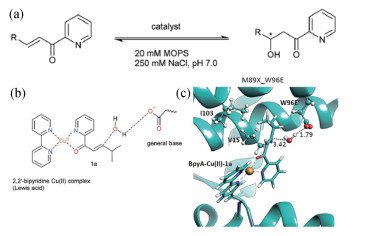
2017年,Roelfes等[32]以LmrR蛋白为模板,通过计算机辅助设计获得了具有水合酶活性的铜蛋白,如图 4所示。首先根据双重激活的反应机理(天冬氨酸或谷氨酸作为广义碱激活水分子,铜离子作为路易斯酸激活共轭双键)计算得到了过渡态构型,然后在不同的位点引入天冬氨酸或谷氨酸作为广义碱,将底物对接到结合位点,最后利用分子动力学模拟评估广义碱、水分子及底物之间的距离是否满足反应的条件。分子动力学结果表明预先设计的广义碱D100与水分子之间不满足反应构型条件,而V15E或W96E突变后的广义碱满足反应构型,实验结果与计算预测结果一致,这表明分子动力学模拟可以作为预测及改造蛋白活性的可靠手段。
图 4

2.2 从头设计新的蛋白骨架来结合金属离子
在含有不同拓扑结构的蛋白中,螺旋束蛋白已经得到了较为透彻的研究,许多数学模型和规律被总结出来[36],通过计算的方法科学家已经可以以较高的成功率获得具有特定对称形式和螺旋数量的螺旋束蛋白的骨架[37, 38]。
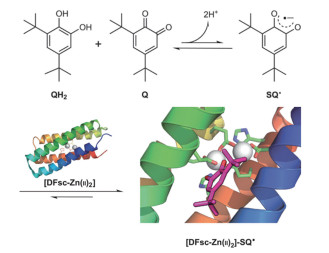
2004年,DeGrado等[39]报道了从头设计得到的异源四聚的螺旋束蛋白,含有双铁中心,可以催化4-氨基苯酚到相应的醌单亚胺的双电子氧化反应。通过在单一催化位点引入双铁中心,反应偏向于双电子氧化还原反应,避免了氧自由基的生成。他们在之前设计得到的非共价自组装形成的DFtet蛋白的基础上引入了底物结合口袋以容纳4-氨基苯酚底物,最终得到了具有中等催化活性的蛋白。催化活性对于底物结合口袋中的甲基取代敏感,证明了设计的特异性。该课题组在金属蛋白设计方向有一系列的工作,主要集中在用螺旋束蛋白的拓扑结构设计具有各种活性的金属蛋白:2009年,获得具有酚氧化活性的双铁蛋白[40];2012年,改造双铁蛋白活性,使其从对苯二酚氧化酶活性变为N-氧化酶活性[41];2014年,设计得到跨膜的锌离子转运螺旋束蛋白[42];2016年,设计得到可以稳定半醌自由基的金属蛋白(如图 5所示)[43]。
图 5

除螺旋束蛋白之外,更加具有普适性的规则和算法被引入来设计不同拓扑结构的蛋白[8, 44~47],与利用已有的蛋白骨架相比,这种从头设计的方法不受蛋白骨架的限制,从设计之初就考虑到金属离子和底物结合位点的各种性质,利于设计得到具有最佳催化构型的蛋白骨架[8]。有研究表明,过渡态-底物结合口袋的完美匹配和催化残基位置的精确摆放是蛋白高催化活性的必要条件[48],因此从头设计蛋白的方法是最有希望得到高催化活性蛋白的方案,这将是未来人工蛋白设计的发展方向。
3. 金属离子的引入——将结合了金属离子的辅因子引入蛋白
从酶催化的角度来看,金属有机催化剂可以看做结合了金属离子的辅因子,由于其独特的催化性质,在C-C键构建等有机合成反应中得到了广泛的研究和应用[49, 50]。将金属有机催化剂引入蛋白的结合口袋,利用蛋白作为反应容器主要有以下优点:一是提高催化效率,反应底物结合到口袋中后,与外界的溶剂分隔开,减少了溶剂的影响,利于反应的进行;二是提高立体选择性,反应物被结合口袋约束成特定的反应构型,减少了非目标反应构型发生的几率,很好地提高了立体选择性。将金属有机催化剂引入到蛋白主要有三种方式:共价结合、非共价结合和配位结合。
3.1 通过共价键或配位结合引入含金属离子的辅因子
半胱氨酸的巯基具有较强的反应活性,适合用于在特定位点利用化学修饰引入小分子配体或金属辅因子。另外也可以利用配体本身与蛋白的非共价相互作用,例如,已知小分子配体A能结合蛋白,将含有金属离子的辅因子共价连接到A上,这样A和辅因子的复合物就结合到了蛋白上,后续可以通过酶的设计(Enzyme Design)优化辅因子和底物结合口袋以增加结合力。
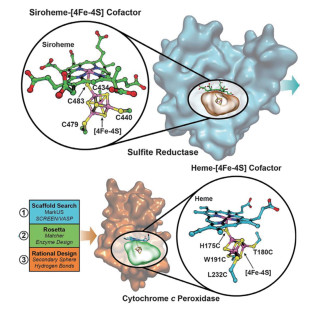
Mirts等[4]通过计算机设计,在细胞色素c过氧化物酶中引入半胱氨酸与[4Fe-4S]簇配位,得到了具有亚硫酸盐还原酶活性的金属蛋白类似物。亚硫酸盐是一种环境污染物,自然界中存在的亚硫酸盐还原酶SiR通过一个6电子转移的还原反应将亚硫酸盐还原为硫化氢,要人工模拟这种多电子转移反应是很困难的。
在SiR中,两个含金属离子的辅因子原血红素和[4Fe-4S]簇立方体通过一个半胱氨酸的硫原子桥联,这种特殊的金属配位中心对于酶活至关重要。设计流程如图 6所示,他们首先在众多蛋白骨架中搜索与SiR活性中心空腔类似的蛋白,确定了以细胞色素c过氧化物酶为骨架,通过rosetta软件中的RosettaMatcher模块引入4个半胱氨酸残基来结合[4Fe-4S]簇立方体,然后通过理性设计周围区域的残基和氢键相互作用,最终得到了催化效率接近天然SiR的人工蛋白。这项工作实现了将两个金属辅因子桥联引入同一个蛋白,对于人工设计催化多电子和多质子转移氧化还原反应的蛋白具有重要意义。
图 6

3.2 通过非共价结合引入含金属离子的辅因子
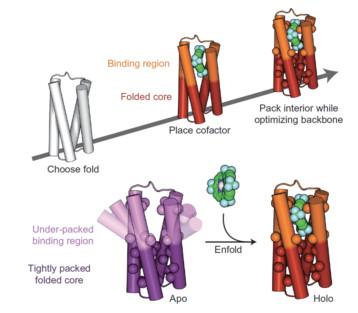
这种方式主要是利用辅因子和蛋白之间的非键相互作用,如范德华力、氢键、疏水相互作用等,实现辅因子配体结合到蛋白口袋中。2017年,DeGrado课题组[51]设计得到的PS1蛋白可以非共价结合一个高度缺电子的卟啉,设计的结构具有亚埃级别的精确度且具有很高的热稳定性。在之前的工作中[52~54],他们开发了计算方法来设计具有原子级别精确度的辅因子结合蛋白,其分步计算策略为:首先通过数学计算生成一个反平行的复绕螺旋结构(coiled coil)蛋白来构建刚性的底物结合位点,其次在蛋白骨架刚性约束的条件下优化残基侧链,重新打包(repack)侧链原子。通过这种从头设计的方法可以得到具有预期三级结构的结合卟啉配体的蛋白,但通常无法达到原子级别的准确度。他们认为,远离底物结合位点之外的其他位点对于底物的结合、催化活性及结构调控都具有很大的影响,因此设计时必须整体考虑所有的位点,整个设计流程如图 7所示。该研究的意义在于作者使用的是真正从头设计的方法,而非采用已有的蛋白骨架;使用了柔性蛋白骨架设计,对所有残基都进行了设计,而不再局限于配体结合位点周围的第二、第三配位层;设计准确度高,不依赖于对众多设计结果的筛选。
图 7

4. 金属蛋白的改造方向
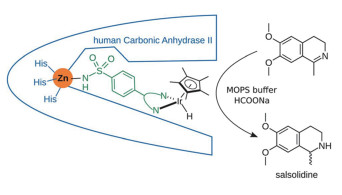
4.1 提高活性
为提高设计得到的金属蛋白的活性,策略之一为通过计算改造增强与辅酶或底物的结合力。2015年,Heinisch等[55]利用RosettaDesign改造了催化不对称氢化还原反应的人工金属酶[(η5-Cp*)Ir(pico)Cl] ⊂WT hCA II (Cp*=Me5C5-)的配体结合口袋,增强了催化活性。在之前的工作中,他们以人源碳酸酐酶(hCA)为蛋白骨架,利用苯磺酰胺高亲和地结合hCA中锌离子的性质,在苯磺酰胺中引入了一个二配位的配体化合物来结合金属有机催化剂(如图 8所示),成功构建得到了可以催化不对称氢化还原反应的金属酶[56]。通过测试不同的配体,ee值最高达到68%。为了进一步提高ee值和催化效率,选择了配体和Ir配合物周围的结合口袋的残基,利用RosettaDesign进行优化,最终将底物的Kd值从21nmol/L提高到了0.33nmol/L,ee值提高到96%,TON从15提高到100。该研究证明了计算机辅助蛋白设计的方法可以有效提高酶中的非天然配体对蛋白的结合能力,进而提高催化活性和对映选择性[57, 58]。
图 8

4.2 产生新的活性
一方面,基于酶催化的机理改造底物结合口袋的残基可以改变底物特异性或者获得具有新颖催化活性的蛋白,如近期Li等[59]利用rosetta软件改造了天冬氨酸酶的活性位点,使其获得了催化不对称氨加成到丙烯酸生成β-氨基酸产物的活性,并成功用于工业级的大规模生物发酵,实现了300g/L的底物浓度下,99%的转化率及>99%位点选择性和对映选择性;另一方面,改变与金属离子配位的氨基酸、改变底物与金属离子配位的方式,甚至改变第二配位层的残基都可以改变金属蛋白原有的催化活性。
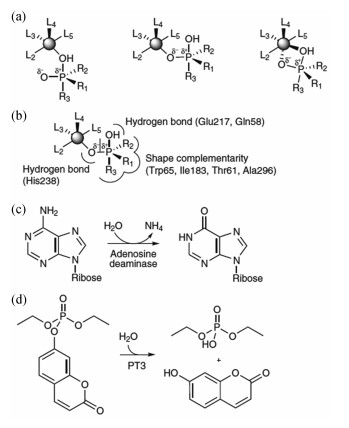
2012年,Khare等[60]开发了新的计算方法用于改造小鼠腺苷脱氨酶的活性位点,得到了具有新的活性的金属蛋白,可以催化有机磷酯键的水解,用于降解有机磷杀虫剂和神经毒剂等,如图 9所示。首先根据对氧磷水解的量子化学计算结果定义了几种不同的反应过渡态构型及锌离子的三角双锥型配位方式,同时引入氢键和形状匹配,然后在蛋白骨架数据库中搜索可能符合要求的蛋白,RosettaDesign优化周围的残基,得到了具有中等催化活性(kcat/Km=4 L·mol-1·s-1)的蛋白,最终经过定向进化后获得了kcat/Km达到约104 L·mol-1·s-1的蛋白PT3.3。其中,PT3.1的设计模型与晶体结构的蛋白骨架均方根误差(RMSD)为0.65Å,活性位点所在的两个环区域有微小的扰动,但总体并没有影响蛋白骨架及活性位点的侧链构型,证明了这种计算方法的可行性。
图 9

5. 利用定向进化改造金属蛋白
定向进化[61]模拟了生物进化过程中自然选择的过程,在人工条件下加速自然选择,每一轮突变都保留对于目标有益的突变,通过数轮反复迭代的过程,积累有益突变,最终可以得到具有很高催化活性、立体选择性或热稳定性的酶。
酶的理性设计一般指根据已知的酶的结构和催化机理,有选择性地突变特定的氨基酸残基,如突变极性氨基酸以改变侧链的pKa,引入空间位阻较大的氨基酸残基以改变催化反应的立体选择性等,是从基本原理出发的科学的改造方法。与理性设计相反,定向进化是一种工程化的改造思路,甚至不需要知道酶的催化机理,只要是有益的突变就保留,多个有益突变的累加实现了由量变到质变的飞跃。由于生物体本身的复杂性和计算模拟方法本身准确度、当前计算能力的限制,酶的定向进化在实际工业应用中比理性设计更加广泛。结合理性设计的指导,定向进化可以更快地达到酶改造的目标。一般来说,通过计算设计得到的金属蛋白起始活性较低,需要通过定向进化的方式提高活性。
2018年诺贝尔化学奖得主Frances H. Arnold是酶定向进化领域的开创者,近年来在利用定向进化改造金属蛋白方面取得了很多突破性的进展。Arnold课题组以含血红素(Heme)的金属蛋白为改造对象,通过定向进化的方法赋予了蛋白突变体在生物体内构建新型化学键的能力。2013年,他们通过对细胞色素P450的定向进化获得了催化卡宾转移实现烯烃环丙烷化反应的酶[62]。2016年,他们通过定向进化细胞色素c实现了酶催化的C-Si键的合成[63]。在此之前催化C-Si键合成的酶从未被报道过,此工作扩展了生物催化反应的化学空间,为具有对映选择性的含硅有机物合成提供了环境友好的高效途径。后续在2017年,利用相似的定向进化策略,Arnold等报道了手性C-B键有机物的酶催化合成[64]以及金属-氧介导的烯烃反马尔科夫尼科夫氧化反应的酶催化合成[65]。2018年,他们通过对血红素蛋白进行定向进化得到了催化卡宾加成到不饱和碳碳双键生成具有高度环张力的手性双环丁烷的酶[66]。
上述很多反应在有机合成中有重要应用,通过定向进化获得具有特定反应活性的酶应用在工业生产中,可以缩短反应时间,提高区域和立体选择性,提高酶对溶剂的耐受性,提高酶的热稳定性,降低生产成本,减少污染,甚至得到无法用常规有机化学合成得到的特殊反应,具有重要应用价值。
6. 结语
研究者在计算机辅助的金属蛋白设计和改造领域取得了众多进展,但该领域也存在很多问题和挑战。(1)设计成功率较低,通常数十条设计得到的蛋白序列中才有一个真正有活性的蛋白;(2)设计得到的酶活性较低,需要通过定向进化等手段才能达到与天然酶活性媲美的程度;(3)目前对金属蛋白的设计主要集中在从天然的蛋白骨架构型、残基配位规律中学习,模拟天然金属蛋白的活性位点,无法人工设计具有指定活性的金属蛋白,根据新的催化机理设计蛋白仍非常困难,成功率较低;(4)目前的设计很难同时兼顾金属离子结合位点、反应底物及过渡态构型,只能重点考虑某一项,而构造原子级别精确度的活性位点结构是高催化活性的基础,必须从整体考虑;(5)不同的金属离子结合位点性质不同,需要更加准确可靠或特殊的力场参数或打分函数进行描述,以提高设计的准确性。
未来本领域可能的发展方向:(1)利用非天然氨基酸进行金属蛋白的设计,目前的工作已经可以设计得到原子级别精确度的金属离子结合位点[29],如果将金属离子催化位点和底物结合位点的特征考虑进去,可以得到具有指定催化活性的金属蛋白,将具有很好的应用前景;(2)相比于从已有的蛋白骨架上构建金属离子活性中心,从头设计蛋白时可以根据指定构型的活性中心进行设计,不局限于已有的金属离子结合模式[8];随着从头设计蛋白准确度的提高,未来将可以从头设计具有指定活性的金属蛋白,这也是蛋白设计领域的重要目标;(3)机器学习、深度学习等人工智能方法[67~69]在蛋白折叠和设计领域中的应用可能会带来人工金属蛋白设计的重大突破。
-
-
[1]
X Lu, C Wang, B Liu. Fish Shellfish Immunol., 2015, 42(1):58~65. doi: 10.1016/j.fsi.2014.10.027
-
[2]
T J Erb, J Zarzycki. Curr. Opin. Biotechnol., 2018, 49:100~107. doi: 10.1016/j.copbio.2017.07.017
-
[3]
K D Jackson, R Durandis, M J Vergne. Int. J. Mol. Sci., 2018, 19(8):2367. doi: 10.3390/ijms19082367
-
[4]
E N Mirts, I D Petrik, P Hosseinzadeh et al. Science, 2018, 361(6407):1098~1101. doi: 10.1126/science.aat8474
-
[5]
A D Pearson, J H Mills, Y Song et al. Science, 2015, 347(6224):863~867. doi: 10.1126/science.aaa2424
-
[6]
A Chevalier, D A Silva, G J Rocklin et al. Nature, 2017, 550:74~79. doi: 10.1038/nature23912
-
[7]
D Rothlisberger, O Khersonsky, A M Wollacott et al. Nature, 2008, 453(7192):190~195. doi: 10.1038/nature06879
-
[8]
E Marcos, B Basanta, T M Chidyausiku et al. Science, 2017, 355(6321):201~206. doi: 10.1126/science.aah7389
-
[9]
C E Tinberg, S D Khare, J Dou et al. Nature, 2013, 501(7466):212~216. doi: 10.1038/nature12443
-
[10]
K T O'Neil, W F DeGrado. Science, 1990, 250(4981):646~651. doi: 10.1126/science.2237415
-
[11]
H W Hellinga, J P Caradonna, F M Richards. J. Mol. Biol., 1991, 222(3):787~803. doi: 10.1016/0022-2836(91)90511-4
-
[12]
N D Clarke, S M Yuan. Proteins, 1995, 23(2):256~263.
-
[13]
A Leaver-Fay, M Tyka, S M Lewis et al. Methods Enzymol., 2011, 487:545~574. doi: 10.1016/B978-0-12-381270-4.00019-6
-
[14]
F Richter, A Leaver-Fay, S D Khare et al. PLoS One, 2011, 6(5):e19230. doi: 10.1371/journal.pone.0019230
-
[15]
D Hilvert. Annu. Rev. Biochem., 2013, 82:447~470. doi: 10.1146/annurev-biochem-072611-101825
-
[16]
G R Nosrati, K N Houk. Protein Sci., 2012, 21(5):697~706. doi: 10.1002/pro.2055
-
[17]
I V Korendovych, D W Kulp, Y Wu et al. PNAS, 2011, 108(17):6823~6827. doi: 10.1073/pnas.1018191108
-
[18]
L Zhou, M Bosscher, C Zhang et al. Nat. Chem., 2014, 6(3):236~241. doi: 10.1038/nchem.1856
-
[19]
E Bozkurt, M A S Perez, R Hovius et al. J. Am. Chem. Soc., 2018, 140(13):4517~4521. doi: 10.1021/jacs.7b10660
-
[20]
M Klemba, K H Gardner, S Marino et al. Nat. Struct. Biol., 1995, 2:368~373. doi: 10.1038/nsb0595-368
-
[21]
C C Liu, P G Schultz. Annu. Rev. Biochem., 2010, 79:413~444. doi: 10.1146/annurev.biochem.052308.105824
-
[22]
L Wang, A Brock, B Herberich et al. Science, 2001, 292(5516):498~500. doi: 10.1126/science.1060077
-
[23]
I Drienovska, C Mayer, C Dulson et al. Nat. Chem., 2018, 10:946~952. doi: 10.1038/s41557-018-0082-z
-
[24]
J Xie, W Liu, P G Schultz. Angew. Chem. Int. Ed., 2007, 46(48):9239~9242. doi: 10.1002/(ISSN)1521-3773
-
[25]
X Liu, J Li, C Hu et al. Angew. Chem. Int. Ed., 2013, 52(18):4805~4809. doi: 10.1002/anie.v52.18
-
[26]
X Liu, J Li, J Dong et al. Angew. Chem. Int. Ed., 2012, 51(41):10261~10265. doi: 10.1002/anie.201204962
-
[27]
X Liu, Y Yu, C Hu et al. Angew. Chem. Int. Ed., 2012, 51(18):4312~4316. doi: 10.1002/anie.201108756
-
[28]
H S Lee, P G Schultz. J. Am. Chem. Soc., 2008, 130(40):13194~13195. doi: 10.1021/ja804653f
-
[29]
J H Mills, S D Khare, J M Bolduc et al. J. Am. Chem. Soc., 2013, 135(36):13393~13399. doi: 10.1021/ja403503m
-
[30]
J H Mills, W Sheffler, M E Ener et al. PNAS, 2016, 113(52):15012~15017. doi: 10.1073/pnas.1600188113
-
[31]
X Luo, T S Wang, Y Zhang et al. Cell. Chem. Biol., 2016, 23(9):1098~1102. doi: 10.1016/j.chembiol.2016.08.007
-
[32]
I Drienovska, L Alonso-Cotchico, P Vidossich et al. Chem. Sci., 2017, 8(10):7228~7235. doi: 10.1039/C7SC03477F
-
[33]
I Drienovska, A Rioz-Martinez, A Draksharapu et al. Chem. Sci., 2015, 6(1):770~776. doi: 10.1039/C4SC01525H
-
[34]
J Bos, W R Browne, A J Driessen et al. J. Am. Chem. Soc., 2015, 137(31):9796~9799. doi: 10.1021/jacs.5b05790
-
[35]
J Bos, F Fusetti, A J Driessen et al. Angew. Chem. Int. Ed., 2012, 51(30):7472~7475. doi: 10.1002/anie.201202070
-
[36]
G Grigoryan, W F Degrado. J. Mol. Biol., 2011, 405(4):1079~1100. doi: 10.1016/j.jmb.2010.08.058
-
[37]
N R Zaccai, B Chi, A R Thomson et al. Nat. Chem. Biol., 2011, 7(12):935~941. doi: 10.1038/nchembio.692
-
[38]
F Lapenta, J Aupic, Z Strmsek et al. Chem. Soc. Rev., 2018, 47(10):3530~3542. doi: 10.1039/C7CS00822H
-
[39]
J Kaplan, W F DeGrado. PNAS, 2004, 101(32):11566~11570. doi: 10.1073/pnas.0404387101
-
[40]
M Faiella, C Andreozzi, R T de Rosales et al. Nat. Chem. Biol., 2009, 5(12):882~884. doi: 10.1038/nchembio.257
-
[41]
A J Reig, M M Pires, R A Snyder et al. Nat. Chem., 2012, 4(11):900~906. doi: 10.1038/nchem.1454
-
[42]
N H Joh, T Wang, M P Bhate et al. Science, 2014, 346(6216):1520~1524. doi: 10.1126/science.1261172
-
[43]
G Ulas, T Lemmin, Y Wu et al. Nat. Chem., 2016, 8(4):354~359. doi: 10.1038/nchem.2453
-
[44]
P S Huang, K Feldmeier, F Parmeggiani et al. Nat. Chem. Biol., 2016, 12(1):29~34. doi: 10.1038/nchembio.1966
-
[45]
Y R Lin, N Koga, R Tatsumi-Koga et al. PNAS, 2015, 112(40):E5478~E5485. doi: 10.1073/pnas.1509508112
-
[46]
T J Brunette, F Parmeggiani, P-S Huang et al. Nature, 2015, 528(7583):580~584. doi: 10.1038/nature16162
-
[47]
B Dang, H Wu, V K Mulligan et al. PNAS, 2017, 114(41):10852~10857. doi: 10.1073/pnas.1710695114
-
[48]
R Blomberg, H Kries, D M Pinkas et al. Nature, 2013, 503(7476):418~421. doi: 10.1038/nature12623
-
[49]
B Albada, N Metzler-Nolte. Chem. Rev., 2016, 116(19):11797~11839. doi: 10.1021/acs.chemrev.6b00166
-
[50]
A H Cherney, N T Kadunce, S E Reisman. Chem. Rev., 2015, 115(17):9587~9652. doi: 10.1021/acs.chemrev.5b00162
-
[51]
N F Polizzi, Y Wu, T Lemmin et al. Nat. Chem., 2017, 9(12):1157~1164. doi: 10.1038/nchem.2846
-
[52]
G M Bender, A Lehmann, H Zou et al. J. Am. Chem. Soc., 2007, 129(35):10732~10740. doi: 10.1021/ja071199j
-
[53]
H C Fry, A Lehmann, J G Saven et al. J. Am. Chem. Soc., 2010, 132(11):3997~4005. doi: 10.1021/ja907407m
-
[54]
H C Fry, A Lehmann, L E Sinks et al. J. Am. Chem. Soc., 2013, 135(37):13914~13926. doi: 10.1021/ja4067404
-
[55]
T Heinisch, M Pellizzoni, M Duerrenberger et al. J. Am. Chem. Soc., 2015, 137(32):10414~10419. doi: 10.1021/jacs.5b06622
-
[56]
F W Monnard, E S Nogueira, T Heinisch et al. Chem. Sci., 2013, 4(8):3269~3274. doi: 10.1039/c3sc51065d
-
[57]
X Feng, J Ambia, K M Chen et al. Nat. Chem. Biol., 2017, 13(7):715~723. doi: 10.1038/nchembio.2371
-
[58]
H J Wijma, R J Floor, S Bjelic et al. Angew. Chem. Int. Ed., 2015, 54(12):3726~3730. doi: 10.1002/anie.201411415
-
[59]
R Li, H J Wijma, L Song et al. Nat. Chem. Biol., 2018:14:664~670.
-
[60]
S D Khare, Y Kipnis, P Greisen et al. Nat. Chem. Biol., 2012, 8(3):294~300. doi: 10.1038/nchembio.777
-
[61]
F H Arnold. Angew. Chem. Int. Ed., 2018, 57(16):4143~4148. doi: 10.1002/anie.201708408
-
[62]
P S Coelho, E M Brustad, A Kannan et al. Science, 2013, 339(6117):307~310. doi: 10.1126/science.1231434
-
[63]
S B J Kan, R D Lewis, K Chen et al. Science, 2016, 354(6315):1048~1051. doi: 10.1126/science.aah6219
-
[64]
S B J Kan, X Huang, Y Gumulya et al. Nature, 2017, 552(7683):132~136. doi: 10.1038/nature24996
-
[65]
S C Hammer, G Kubik, E Watkins et al. Science, 2017, 358(6360):215~218. doi: 10.1126/science.aao1482
-
[66]
K Chen, X Huang, S B J Kan et al. Science, 2018, 360(6384):71~75. doi: 10.1126/science.aar4239
-
[67]
K K Yang, Z Wu, C N Bedbrook et al. Bioinformatics, 2018, 34(15):2642~2648. doi: 10.1093/bioinformatics/bty178
-
[68]
C N Bedbrook, K K Yang, A J Rice et al. PLoS Comput. Biol., 2017, 13(10):e1005786. doi: 10.1371/journal.pcbi.1005786
-
[69]
P A Romero, A Krause, F H Arnold. PNAS, 2013, 110(3):E193~E201. doi: 10.1073/pnas.1215251110
-
[1]
-

图式 1 具有金属离子螯合性质的非天然氨基酸及其类似物的结构式
Scheme 1 Chemical structure of metal-chelating UAA and analogues
-

 下载:
下载:

 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 18
- 文章访问数: 766
- HTML全文浏览量: 234
