引用本文:
惠鹏, 杨蓉, 邓七九, 燕映霖, 许云华. 金属氧化物用于锂硫电池硫正极材料改性的研究进展[J]. 化学通报,
2019, 82(11): 982-988.

Citation: Hui Peng, Yang Rong, Deng Qijiu, Yan Yinglin, Xu Yunhua. Research Progress of Metal Oxides for Modification of Sulfur Cathode in Lithium-Sulfur Batteries[J]. Chemistry, 2019, 82(11): 982-988.
Citation: Hui Peng, Yang Rong, Deng Qijiu, Yan Yinglin, Xu Yunhua. Research Progress of Metal Oxides for Modification of Sulfur Cathode in Lithium-Sulfur Batteries[J]. Chemistry, 2019, 82(11): 982-988.
金属氧化物用于锂硫电池硫正极材料改性的研究进展
摘要:
锂硫电池因其能量密度高、原料丰富和价格低廉等优势而被认为是下一代的重要储能器件。但是,锂硫电池的发展仍面临诸多问题,包括多硫化物的穿梭效应、单质硫的导电性差、充电过程中硫体积膨胀导致的库仑效率差、容量快速衰减以及锂负极的腐蚀等。近年来,金属氧化物由于具有可吸附多硫化物、提高多硫化物之间的相互转化能力、形成3D形态纳米级结构及对主体材料与多硫化物之间的结合能发挥着关键作用等优点在锂硫电池正极材料的改性方面得到广泛应用。本文综述了多类金属氧化物(过渡金属氧化物、二元及多元金属氧化物、其他金属氧化物)在锂硫电池正极复合材料改性中的研究进展,并对金属氧化物在锂硫电池中的应用前景进行了展望。
English
Research Progress of Metal Oxides for Modification of Sulfur Cathode in Lithium-Sulfur Batteries
Abstract:
Lithium-sulfur batteries are considered to be the next generation of energy storage devices due to their high energy density, abundant raw materials and low price. However, the development of lithium-sulfur batteries still faces many problems, including the shuttle effect of polysulfide, the poor conductivity of elemental sulfur, the poor coulombic efficiency caused by sulfur volume expansion during charging, the rapid decay of capacity and the corrosion of lithium anode. In recent years, metal oxides have been widely used in the modification of positive electrodes of lithium-sulfur batteries because of their ability to adsorb polysulfides, improving the mutual conversion ability between polysulfides, forming nanoscale structure with a 3D morphology, and playing a key role in the combination of host material and polysulfide. In this paper, the research progress in various metal oxides (transition metal oxides, binary and multi-metal oxides, other metal oxides) for the modification of lithium-sulfur battery cathode composites is reviewed. The application prospect of metal oxides in lithium-sulfur batteries is prospected.
-
Key words:
- Lithium-sulfur battery
- / Metal oxide
- / Cathode composite
- / Electrochemistry
- / Energy storage mechanism
-
随着能源结构变革,化石能源的不断消耗引发人类对绿色清洁能源的广泛关注,这极大地刺激了新一代储能器件的开发应用。近年来,锂硫电池因具有高比容量(1675mAh/g)和高能量密度(2600Wh/kg,是锂离子电池(≈360Wh/kg)的7倍左右)以及单质硫(正极活性物质)无毒、成本低、储量丰富等优势[1],在电动汽车、无人机、军用便携式电源、储能系统等领域有着广阔的应用前景。但其发展仍然存在如下问题:(1)硫的导电性差[2];(2)多硫化物在电解质中溶解引起“穿梭效应”[3];(3)在充电过程中硫体积膨胀(约80%)和放电过程中正极上形成的锂枝晶导致库仑效率差,容量衰减快,以及锂金属负极的腐蚀[4~7]。研究初期,人们在正极中添加导电碳材料(主要是为了提高正极的导电性)和氧化物以及采用聚合物电解质解决此类问题,但从世界范围来看,限制锂硫电池发展的关键问题尚未彻底解决。以动力电池为例,其不仅需要有较高的能量密度和较低的成本,还要求出色的安全性、功率密度和循环稳定性等。锂硫电池这些性能指标的实现与其工作原理、关键材料、电池结构、制备工艺和应用模式都有密切的关系。
锂硫电池的工作原理[8~11]为:充电时,Li2S电解生成长链Li2Sn(n=2~8),Li+迁移至负极沉积为金属锂;放电过程中,硫首先锂化形成一系列中间的长链多硫化锂(S8→Li2S8→Li2S6→Li2S4),在进一步锂化时,溶解的长链多硫化物形成短链多硫化物(Li2S4→Li2S2→Li2S),生成固体物质沉淀到电极上,伴随着S-S键的断裂和生成,电能和化学能相互转换。锂硫电池的正极材料主要影响锂硫电池的比能量、比功率和能量转换效率[12]。金属氧化物在硫正极材料改性方面具有独特的优势,表现在:在充放电过程中,金属氧化物能通过物理/化学作用束缚多硫化物,抑制穿梭效应,容纳硫体积膨胀;其次,提高多硫化物之间的相互转化能力[13];最后,提供更多的活性位点吸附多硫化物。金属氧化物中极性表面的氧阴离子和金属之间的强结合作用导致大多数金属氧化物不溶于有机溶剂。另外,金属氧化物的内在缺陷和独特能带结构表现出更丰富的极性活性位点用以吸附多硫化物,使得锂硫电池能量密度显著提高。本文主要介绍金属氧化物(过渡金属氧化物、二元及多元金属氧化物、其他金属氧化物)在锂硫电池复合材料应用中的研究进展。
1. 过渡金属氧化物
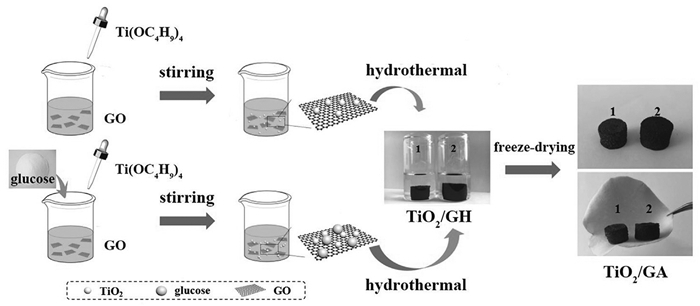
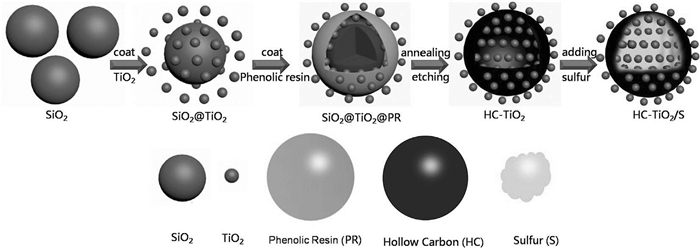
钛氧化物存在自然界中,并具有多种晶型,TiO2就是其中一种,它可来源于锐钛矿(α-TiO2)、金红石(β-TiO2)或板钛矿(γ-TiO2)[14]。由于TiO2具有极性表面,也常以各种形式应用于锂硫电池中。Wang等[15]通过一锅法化学合成工艺制备了轻质三维分层多孔TiO2/石墨烯气凝胶复合材料(3D TiO2/GA)(如图 1)。其在0.2、0.5、1和2 C时初始放电容量分别为1273、1178、1039和1011 mAh/g。2C下500次循环后容量保持率超过50%,库仑效率约为100%。Yao等[16]制备了中空碳球/TiO2/S复合材料(HC/TiO2/S)作为正极(硫含量为67(wt)%)(如图 2)。HC-TiO2/S正极增强的电化学特性源于空心碳球和TiO2的协同效应。中空碳球弥补了导电性差的不足且容纳硫体积膨胀;TiO2和多硫化物的强相互作用可抑制其扩散。硫正极的高理论比容量(1675mAh/g)归因于反应S8+16Li↔8Li2S。类似地,Chen等[17]制备了一种碳纳米管(CNTs)与TiO2-B纳米管(TNTs)交织组成三维网络复合材料(TNT@CNT)(硫负载量为3.2mg/cm2)作为正极硫主体。Ma等[18]先采用改进的静电纺丝技术和热处理制备了TiO2纳米纤维,再用熔融扩散法将硫与TiO2纳米纤维结合制得S-TiO2复合材料。
图 1
图 2

还有利用离子液体(ILs)的多种非共价相互作用(如静电相互作用、氢键和π-π堆积)来合成金属氧化物的策略[19]。Sun等[20]通过一步水热法合成氯化1-羧乙基-3-甲基咪唑鎓,经非共价相互作用引发TiO2纳米颗粒的自组装,进而制备了具有弹性介孔结构的TiO2亚微球(MS TiO2),其孔体积为0.792cm3/g,比表面积为291.08m2/g。此结构具有大机械强度和足够的电子/离子传输通道,实现了主体材料与多硫化物之间有效接触,3D中孔结构能够束缚中间产物容纳硫体积膨胀。
Qian等[21]采用静电纺丝技术和热处理制备介孔TiO2纳米管(MTDNTs)作为正极硫主体,通过将MTDNTs与S/CS2溶液混合来制备MTDNTs/S复合正极。TiO2纳米管能够封装硫,其中空结构有利于Li+的快速传输。Zhao等[22]通过两步正极氧化法制备高度有序的TiO2纳米管阵列,再通过电化学沉积沿着纳米管壁生长聚吡咯(PPy)层,在负载硫后热处理获得具有同轴非均相结构的纳米管阵列正极。用弹性PPy修饰的TiO2纳米管展现两者的协同作用,为锂离子扩散和容纳硫体积膨胀提供高度有序的导电框架,且其蓬松的结构更易形成C-S键。在制备纳米线阵列方面,Liu等[23]采用脱合金方法制备的TiO2表现出由纳米线和均匀纳米多孔组成的海胆样结构。该结构使硫和主体材料充分接触,起到捕获多硫化物中间体并抑制穿梭效应的作用。
上述复合材料均具有以下特点:首先,利用碳的良好导电性降低电阻,提高复合材料的导电能力;其次,掺入的TiO2可物理捕获多硫化物和容纳硫体积膨胀,提供对多硫化物的化学吸附位点与Li2Sn相互作用产生Ti-S键,并且TiO2具有一定的孔道结构,可促进离子传输和硫扩散;最后,TiO2与复合孔道结构产生协同作用,加速游离电子并促使锂离子转移到导电性差的硫中。
研究者除了将TiO2应用于硫正极中有效捕获多硫化物之外,还将Ti4O7用作锂硫电池正极添加剂(具有导电性的Magneli相Ti4O7能够与硫高效结合),达到捕获多硫化物的目的[24, 25]。目前人们已经开发出许多Ti4O7颗粒的制备方法,包括退火活化TiO2和Ti的混合物[26]、用碳或聚乙烯醇[27, 28]加热TiO2、用锆还原锐钛矿TiO2[29]、在NH3气氛中还原金红石TiO2[30]等。
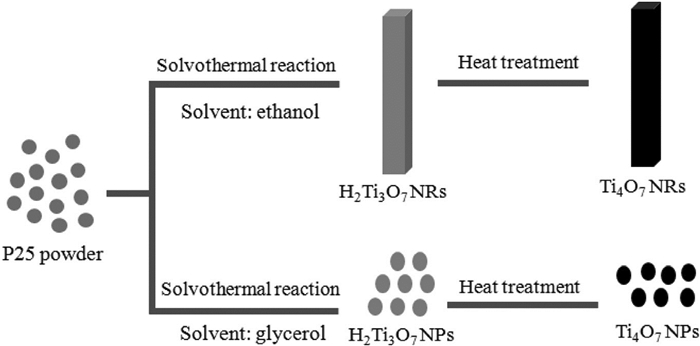
Zhang等[31]通过溶剂热辅助热处理合成了一维Ti4O7纳米棒(1D Ti4O7NRs)和三维Ti4O7纳米粒子(3D Ti4O7NPs)(如图 3)。与Ti4O7纳米颗粒/硫电极相比,添加Ti4O7NRs的硫正极具有良好的比容量(在2C下为590mAh/g)和循环稳定性(在300次循环后比容量为580mAh/g)。优异的电化学性能归因于锂-硫氧化还原反应中较高的催化活性、较低的电荷转移电阻(86.13Ω)和较大的锂离子扩散系数(8.36×10-13cm2/s)。
图 3

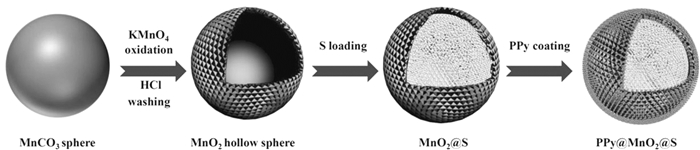
无论MnO2是形成纤维还是多孔球体,它的孔道结构既能通过物理化学作用吸附多硫化物,又能提供空间容纳硫体积膨胀,且纳米硫在MnO2孔道中的均匀分布有助于Li+快速储存和能量快速输送。Li等[32]设计合成了具有双核-壳结构的PPy@MnO2@S球(见图 4)。位于复合球体内部的硫纳米颗粒(约80nm)有助于快速Li +储存;MnO2空心球不仅可以提供足够的内部空间来缓解硫的体积膨胀,而且可以通过结构限制和化学吸附的协同作用有效缓解多硫化物的溶解;另外,第二层壳由PPy纳米颗粒组成,其作为导电框架并产生足够的导电通路。
图 4

Dang等[33]制备了新型形貌的介孔MnO2纤维作为锂硫电池主体材料。MOF/S复合电极在0.1C下初始容量高达1015mAh/g,200次循环后容量保持在815mAh/g,在1C下也表现出优异的循环性能,300次循环后容量保持在796mAh/g。
上述报道是利用MnO2在形成纤维和多孔球体时产生的孔道结构特点进行实验设计,也有研究者通过将MnO2掺杂到复合材料中与其协同作用进而提升电化学性能。例如,Chen等[34]通过一种物理和化学双重封装法,将在氮掺杂的中空多孔碳纳米球(NHCSs@MnO2)上生长的氧化锰纳米片引入硫正极达到捕获多硫化物的目的。形成的纳米球可构建导电网络,降低电子、离子的转移电阻,束缚活性物质,容纳硫体积膨胀,通过协同作用防止多硫化物溶解和扩散。HCSs中氮原子的引入可提供用于电荷快速传输的活性位点。类似地,Zhu等[35]先静电纺丝后热处理制得MnO纳米颗粒(MnO NPs)修饰的碳纳米纤维(CNF)复合材料(CNF-MnO)。在CNF中均匀分布的MnO可通过化学吸附作用有效限制穿梭效应,而CNF和MnO NPs形成新的3D导电结构使锂离子快速传导。
另外,Sun等[36]通过两步涂层法将石墨烯涂覆在硫正极表面,再将MnO2原位生长到石墨烯(MnO2@GR)上以形成复合双层电极。充放电时石墨烯和MnO2与可溶性多硫化物之间具有强化学作用。在0.5C时,初始比容量为1395mAh/g,库仑效率接近100%。Liu等[37]提出了一种新的物理和化学方法捕获多硫化物,将碳支架(CMK-3)和极性涂层(MnO2)引入到硫正极形成MnO2@CMK/S复合材料(硫负载量为73.4(wt)%,MnO2为6.6(wt)%)。CMK-3提供导电路径,MnO2通过化学作用结合多硫化物。除此之外,Wen等[38]通过浸渍法将纳米Mn3O4颗粒原位沉积到类似羊毛的CNTs微球的丰富孔道中形成新型Mn3O4-CNTs微球,再用熔融扩散法将S与Mn3O4-CNTs微球结合。在这种混合结构中,CNTs微球能物理束缚多硫化物,而Mn3O4纳米晶体可化学结合多硫化物。
氧化钼具有独特的二维层状结构和高插入/脱嵌的可逆性,已经有几种形态的钼氧化物(MoO2、MoO3、MoOx)应用于锂硫电池电极材料的报道。Chen等[39]用原位热还原法制备MoO2有序介孔碳CMK-3(M-OMC)混合物作为硫正极主体。M-OMC-S(硫含量约为67%)正极的优异电化学性能归因于高导电率(190S/cm)的MoO2附着在具有导电性的介孔碳骨架表面,其孔道结构可储存活性物质,构建电子快速传输通道并促进多硫化物的转化以缓解穿梭效应。Fang等[40]在碳纤维泡沫电极中加入MoC@MoOx材料可使电化学性能显着提高,复合电极具有高比容量和良好的循环稳定性。
在Fe3O4应用于锂硫电池的研究中,可首先合成铁基金属有机骨架(Fe-MOF),再将其碳化获得Fe3O4/C复合物。所制备的Fe3O4/C复合材料显示出诸多优点:(1)Fe3O4化学吸附多硫化物,抑制穿梭效应;(2)Fe3O4/C含有丰富的碳可提高导电性;(3)Fe3O4/C复合材料中空结构含有较大的空间可以储硫,缓解锂化过程中的体积膨胀;(4)Fe3O4/C复合材料的多孔结构可为多硫化物的吸附提供更多的活性位点。Fan等[41]通过一步水热法合成Fe-MOF衍生的枣核状Fe3O4/C复合物用作锂硫电池的正极硫主体。Fe3O4可通过路易斯酸碱相互作用固定多硫化物;另一方面,复合材料的多孔中空结构不仅可以提供足够的储硫空间,而且还提供足够的间隙以适应锂化过程中的体积膨胀。Chen等[42]先合成六角形二芳基Fe-MOFs模板,再将其在N2气氛下热处理获得具有八面体结构的Fe3O4/C纳米复合材料。Fe3O4/C八面体纳米结构结合了Fe3O4与碳基体的双重优势,提供高锂储存能力和长循环稳定性,多孔纳米结构提高了Li+扩散速率并在充放电过程中克服了大体积膨胀/收缩和严重的颗粒聚集,而掺杂无定形碳形成碳骨架使得电导率提高。
Yang等[43]用ZnO纳米晶须为模板,通过化学气相沉积(CVD)再进行部分原位氢蚀刻工艺合成氧化锌嵌入式四足形碳壳(TCS/ZnO)复合材料。ZnO纳米粒子锚定多硫化物并为其氧化还原提供有效的表面积以及低电阻,中空TCS能容纳硫体积变化。通过熔体扩散法将硫负载于TCS/ZnO复合材料,所制备的TCS/ZnO/S正极材料(ZnO含量为6%)初始比容量为1284mAh/g,库仑效率为99.5%,0.2C下100次循环后容量为815mAh/g。
2. 二元及多元金属氧化物
目前报道的二元及多元材料有Mg0.6Ni0.4O、Li4Ti5O12、LiMn2O4、LixLayTiO3、LiNi0.8Co0.15Al0.05O2等。研究者在改善锂硫电池的性能方面所采取的措施有:(1)碳基材料用作硫正极的主体[44, 45];(2)硫正极涂覆导电聚合物[46];(3)纳米氧化物作为硫正极添加剂[47, 48]。例如,Song等[49]通过溶胶-凝胶法制备Mg0.6Ni0.4O纳米粒子作为Li/S可充电电池的硫正极的电化学惰性添加剂,不仅提高了正极材料的倍率性能,而且提高了电池的充放电容量和循环耐久性。Zhang等[50]通过自蔓延高温合成(SHS)制备纳米Mg0.6Ni0.4O,接着用硬模板法制备介孔碳(MPC),再用化学原位沉积负载硫形成纳米镁镍氧化物-介孔碳/硫复合材料(Mg0.6Ni0.4O-MPC/S)。其中多孔结构的MPC(表面积高达1001.2m2/g)既能吸收多硫化物又能增强导电性,且为Li+提供传输通道。该正极材料在0.1C时初始容量为1360mAh/g,100次循环后容量为695mAh/g[51]。
尖晶石LiMn2O4由于其结构稳定、价格低廉、锂离子传导性好以及可捕获多硫化物的Mn原子活性位点多而被引入锂硫电池正极中。Ji等[52]用熔融扩散法制备了S/LiMn2O4复合材料,其对Li2S6有强吸附作用。吸附机理为:LiMn2O4表面的部分Mn4+被还原并转化为Mn3+和Mn2+,同时,一些S62-被氧化并转化为Li2S6-x(1<x<6)吸附在S/LiMn2O4电极上。S/LiMn2O4电极在0.5C下第1和第300次循环时容量分别为924.9和728.0 mAh/g,超过S/乙炔黑电极的容量(722.1和518.9 mAh/g),表明尖晶石LiMn2O4可以显著提高锂硫电池的电化学性能。
实现长寿命是锂硫电池成为最有前途的下一代储能设备之一的关键。然而,多硫化物的穿梭效应和低氧化还原反应动力学导致活性材料的快速损失、差的速率性能和严重的自放电。为了解决上述问题,Shi等[53]以LiNi0.8Co0.15Al0.05O2层状三元正极材料(NCA)作为捕获多硫化物的主体并同时加速多硫化物的转化。他们证实,NCA的(104)、(003)和(110)晶面通过形成Li-O和Co-S键强烈吸附Li2S6;同时,NCA的极性表面促进了多硫化物的氧化还原反应;此外,NCA可有效改善锂硫电池的静态稳定性并抑制其自放电行为。因此,在1C下循环500次后,锂硫电池实现了755.4mAh/g的高放电容量和0.02%/圈的低容量衰减率。这项工作提供了一种方便有效的方法来提高锂硫电池的实际应用能力。
3. 其他金属氧化物
纳米Al2O3既能吸附多硫化物又能增强离子传导,但由于多孔碳的疏水性和传质阻力,传统方法(如浸渍、沉积等)很难使纳米Al2O3均匀进入孔隙。因此,Wu等[54]用有机吸附和原位热还原方法合成了一种含有碳涂层的独特γ-Al2O3@C核-壳微球,其核-壳结构可包封多硫化物,γ-Al2O3@C的大空间可容纳硫体积膨胀,γ-Al2O3和C之间具有协同作用。Ousmane等[55]通过双模板NaCl-KCl共晶和纳米CaCO3热解葡萄糖-尿素树脂填充MIL-53来制备掺有γ-Al2O3的取向大孔-碳(OMC)。葡萄糖-尿素树脂和NaCl-KCl共晶填充MIL-53中的孔道,以避免在热解过程中孔道坍塌和融合;纳米CaCO3留在隧道外,以防止形成密封的孔道。合成的OMC表现出优异的多硫化物吸收能力,这归因于其超高比表面积和共存活性O和N位点对多硫化物的吸附。所得硫电极显示出高放电容量、长循环寿命和优异的倍率性能。其在0.05C下的初始容量为1626mAh/g,在10C下的比容量高达430mAh/g。经0.2C下300次循环后,容量保持在850mAh/g。
Ma等[56]通过水热法和高温固态反应合成高S负载率的Al2O3掺杂ZnO(AZO)修饰的碳纳米管(AZO@S/CNT)复合材料(硫负载率为75(wt)%)。Al2O3掺杂ZnO后,带隙减小导电性增强,促进了氧化还原反应,且其与多硫化物产生化学作用,抑制了穿梭效应。
制备S/聚合物复合材料的方法包括:将导电聚合物和升华硫热处理混合后再涂覆,其中形成有机硫键[57~59];将升华硫掺入聚合物纳米管中[60, 61];用硫原位沉积导电聚合物[62]。Yermukhambetova等[63]将纳米Al2O3湿法球磨,再经热处理制得S/聚苯胺复合材料,其具有小粒径、多孔结构和高比表面积与孔体积等特点。Al2O3既能吸收电解质又能减缓穿梭效应。
除此之外,还可将SnO2用于锂硫电池正极复合材料。Liu等[64]通过熔融扩散法制备含有SnO2、还原氧化石墨烯(rGO)和CNT的复合材料(SnO2@rGO/CNTs/S)应用于锂硫电池的正极。其中,SnO2、rGO和CNT之间存在协同效应:SnO2能有效吸附多硫化物和减少穿梭效应,但其导电性差;rGO由于大比表面积和良好导电性用作SnO2模板以避免其聚集并增加SnO2反应位点;CNT和rGO构建复合材料的导电网络,加速电子和离子转移,提高活性材料的利用率,改善导电性和机械性能。
Wu等[65]用一锅法制备负载在硫掺杂rGO上的SnO2量子点(表示为SnO2 QDs@S-rGO),其中SnCl2和二硫化苄(BDS)用作GO的还原剂。SnO2 QDs与S-rGO之间具有协同作用:QDs在电化学反应中保持微小尺寸进而减轻体积膨胀,其均匀分散为锂嵌入和脱嵌提供了大量活性位点和空隙;rGO用作导电基体,提升结构稳定性;S-rGO引入的缺陷有利于电荷转移和锂离子扩散。
Zhang等[66]通过两步水热法先制备MoS2纳米片,再将SnO2纳米粒子均匀涂覆在MoS2纳米片上制得分级MoS2/SnO2纳米复合材料,并将其用作锂硫电池正极。它在正极的电化学反应为:
$\mathrm{SnO}_{2}+4 \mathrm{Li}^{+}+4 \mathrm{e}^{-}=\mathrm{Sn}+2 \mathrm{Li}_{2} \mathrm{O} $
(1) $\mathrm{MoS}_{2}+x \mathrm{Li}^{+}+x \mathrm{e}^{-}=\mathrm{Li}_{x} \mathrm{MoS}_{2} $
(2) $\mathrm{Li}_{x} \mathrm{MoS}_{2}+(4-x)\left(\mathrm{Li}^{+}+\mathrm{e}^{-}\right)=\mathrm{Mo}+2 \mathrm{Li}_{2} \mathrm{S} $
(3) $\operatorname{Sn}+x\left(\mathrm{Li}^{+}+\mathrm{e}^{-}\right)=\mathrm{Li}_{x} \mathrm{S}_{n}(0 \leqslant x \leqslant 4.4) $
(4) MoS2和SnO2之间存在协同效应,负载的SnO2纳米颗粒抑制多硫化锂,金属钼颗粒可增强复合材料导电性。
随后,Gao等[67]通过类似方法制得CoMoS3.13/SnO2纳米复合材料。SnO2纳米粒子壳能抑制多硫化锂扩散,避免活性材料的损失,使得锂硫电池的电化学性能在应用中得到较大提升。Xiao等[68]通过溶剂热反应合成CeO2网状碳纳米管(CeO2@CNT),再将硫用化学法负载到CeO2@CNT中,使得电化学性能大大提高。具有3D高导电多孔网络的碳纳米管为电子、离子转移提供最佳通道;而极性CeO2化学键合多硫化物防止其溶解于电解质。Li等[69]通过溶胶-凝胶工艺原位合成了不同含量氧化钕掺杂的碳气凝胶(CA)材料,并用作锂硫电池正极主体。通过钕和硫之间的化学作用将硫锚定在具有互连孔结构的CA中,中孔CA材料和良好分布的Nd2O3使得电化学性能增强。复合Nd2-CA/S正极(硫含量为55.72%)在0.2C时初始放电容量为1327mAh/g,100次循环后容量为1082mAh/g。在0.5C下300次循环后容量为907mAh/g。结果表明,金属氧化物纳米粒子修饰的碳材料可以稳定地固定硫正极,显著提高锂硫电池的电化学性能。
4. 结语
本文综述了金属氧化物用于锂硫电池正极材料的改性研究进展。在改善锂硫电池的电化学性能方面金属氧化物所起的作用包括:(1)对多硫化物进行强烈的化学吸附进而抑制穿梭效应;(2)提高多硫化物之间的相互转化能力;(3)形成的纳米结构提供更多的活性位点吸附多硫化物。
对于锂硫电池的实际应用,除了提供整体解决方案之外,还要注重其他问题的解决,如硫负载量较低、硫与电解质的体积比大、循环过程中质量/体积能量密度的下降等。在电池正极材料中将金属氧化物用于提高硫的负载量还有很长的路要走。另外,必须重视长链多硫化物和短链多硫化物的转化机理研究。金属氧化物中的一部分可以提供电催化作用以介导多硫化物的氧化还原反应。此外,Li2S2到Li2S的固态转换应该高度重视,如果Li2S2全部生成Li2S且两者之间的氧化还原有助于提高锂硫电池的能量密度。在将来的锂硫电池应用研究中,提高活性物质的负载量、限制活性物质的溶解、降低穿梭效应、锂金属负极中锂枝晶问题等仍需要重点考虑。随着对电池结构的进一步优化,相信这些问题在将来都会逐步得到解决。
-
-
[1]
L Carbone, T Coneglian, M Gobet et al. J. Power Sources, 2018, 377: 26~35. doi: 10.1016/j.jpowsour.2017.11.079
-
[2]
Y Zhang, Z Bakenov, Y Zhao et al. Powder Technology, 2013, 235: 248~255. doi: 10.1016/j.powtec.2012.10.023
-
[3]
A Fotouhi, D Auger, L O'Neill et al. Energies, 2017, 10(12): 1937. doi: 10.3390/en10121937
-
[4]
J Ma, Z Fang, Y Yan et al. Adv. Energy Mater., 2015, 5(16): 1500046. doi: 10.1002/aenm.201500046
-
[5]
N Jayaprakash, J Shen, S S Moganty et al. Angew. Chem. Int. Ed., 2011, 50(26): 5904~5908. doi: 10.1002/anie.201100637
-
[6]
F Wu, J Chen, R Chen et al. J. Phys. Chem. C, 2011, 115(13): 6057~6063. doi: 10.1021/jp1114724
-
[7]
Z Wei Seh, W Li, J J Cha et al. Nat. Commun., 2013, 4: 1331. doi: 10.1038/ncomms2327
-
[8]
X Ji, K T Lee, L F Nazar et al. Nat. Mater., 2009, 8(6): 500~506. doi: 10.1038/nmat2460
-
[9]
P G Bruce, S A Freunberger, L J Hardwick et al. Nat. Mater., 2011, 11(1): 19~29. 10.1038/NMAT3237
-
[10]
L Yuan, X Qiu, L Chen et al. J. Power Sources, 2009, 189(1): 127~132. doi: 10.1016/j.jpowsour.2008.10.033
-
[11]
S Walus, C Barchasz, R Bouchet et al. Adv. Energy Mater., 2015, 5: 1500165. doi: 10.1002/aenm.201500165
-
[12]
Y S Su, Y Fu, T Cochell et al. Nat. Commun., 2013, 4(1): 2985. doi: 10.1038/ncomms3985
-
[13]
J H Kim, S Y Choi, M Choi et al. Adv. Energy Mater., 2016, 6(6): 1501902. doi: 10.1002/aenm.201501902
-
[14]
Z Yang, D Choi, S Kerisit et al. J. Power Sources, 2009, 192(2): 588~598. doi: 10.1016/j.jpowsour.2009.02.038
-
[15]
J Wang, C Fu, X Wang et al. Electrochim. Acta, 2018, 292: 568~574. doi: 10.1016/j.electacta.2018.09.109
-
[16]
J Yao, T Mei, Z Cui et al. Chem. Eng. J., 2017, 330: 644~650. doi: 10.1016/j.cej.2017.08.006
-
[17]
A Chen, W Liu, H Hu et al. J. Power Sources, 2018, 400: 23~30. doi: 10.1016/j.jpowsour.2018.08.004
-
[18]
X Ma, B Jin, H Wang et al. J. Electroanal. Chem., 2015, 736: 127~131. doi: 10.1016/j.jelechem.2014.11.007
-
[19]
T L Greaves, C J Drummond. Chem. Soc. Rev., 2013, 42(3): 1096~1120. doi: 10.1039/C2CS35339C
-
[20]
Y Sun, Y Zhao, Y Cui et al. Electrochim. Acta, 2017, 239: 56~64. doi: 10.1016/j.electacta.2017.04.007
-
[21]
X Qian, X Yang, L Jin et al. Mater. Res. Bull., 2017, 95: 402~408. doi: 10.1016/j.materresbull.2017.07.009
-
[22]
Y Zhao, W Zhu, G Z Chen et al. J. Power Sources, 2016, 327: 447~456. doi: 10.1016/j.jpowsour.2016.07.082
-
[23]
N Liu, L Wang, Y Zhao et al. J. Alloys Compd., 2018, 769: 678~685. doi: 10.1016/j.jallcom.2018.08.027
-
[24]
Q Pang, D Kundu, M Cuisinier et al. Nat. Commun., 2014, 5: 4759. doi: 10.1038/ncomms5759
-
[25]
S Yao, S Xue, Y Zhang et al. J. Mater. Sci-Mater. El., 2017, 28(10): 7264~7270. doi: 10.1007/s10854-017-6410-z
-
[26]
A A Gusev, E G Avvakumov, A Z Medvedev et al. Sci. Sintering, 2007, 39(1): 51~57. doi: 10.2298/SOS0701051G
-
[27]
M Toyoda, T Yano, B Tryba et al. Appl. Catal. B, 2009, 88(1/2): 160~164. 10.1016/j.apcatb.2008.09.009
-
[28]
X Zhang, Y Lin, X Zhong et al. J. Mater. Sci-Mater. El., 2016, 27(5): 4861~4865. doi: 10.1007/s10854-016-4368-x
-
[29]
A Kitada, G Hasegawa, Y Kobayashi et al. J. Am. Chem. Soc., 2012, 134(26): 10894~10898. doi: 10.1021/ja302083n
-
[30]
C Tang, D Zhou, Q Zhang et al. Mater. Lett., 2012, 79: 42~44. doi: 10.1016/j.matlet.2012.03.095
-
[31]
Y Zhang, S Yao, R Zhuang et al. J. Alloys Compd., 2017, 729: 1136~1144. doi: 10.1016/j.jallcom.2017.09.252
-
[32]
Y Li, B Shi, W Liu et al. Electrochim. Acta, 2018, 260: 912~920. doi: 10.1016/j.electacta.2017.12.068
-
[33]
R Dang, X Ma, J Liu et al. Int. J. Hydrogen Energy, 2018, 43(41): 18754~18758. doi: 10.1016/j.ijhydene.2018.08.068
-
[34]
M Chen, Q Lu, S Jiang et al. Chem. Eng. J., 2018, 335: 831~842. doi: 10.1016/j.cej.2017.11.039
-
[35]
J Zhu, R Pitcheri, T Kang et al. Ceram. Int., 2018, 44(14): 16837~16843. doi: 10.1016/j.ceramint.2018.06.119
-
[36]
W Sun, X Ou, X Yue et al. Electrochim. Acta, 2016, 207: 198~206. doi: 10.1016/j.electacta.2016.04.135
-
[37]
J Liu, C Wang, B Liu et al. Mater. Lett., 2017, 195: 236~239. doi: 10.1016/j.matlet.2017.02.116
-
[38]
X Wen, K Xiang, Y Zhu et al. Mater. Lett., 2018, 229: 272~276. doi: 10.1016/j.matlet.2018.07.014
-
[39]
Y Chen, S Niu, W Lv et al. Chin. Chem. Lett., 2019, 30(2): 521~524. doi: 10.1016/j.cclet.2018.04.019
-
[40]
R Fang, S Zhao, Z Sun et al. Energy Storage Mater., 2018, 10: 56~61. doi: 10.1016/j.ensm.2017.08.005
-
[41]
L Fan, H Wu, X Wu et al. Electrochim. Acta, 2019, 295: 444~451. doi: 10.1016/j.electacta.2018.08.107
-
[42]
Y Chen, L Zheng, Y Fu et al. RSC Adv., 2016, 6(89): 85917~85923. doi: 10.1039/C6RA19041C
-
[43]
R Yang, H Du, Z Lin et al. Carbon, 2019, 141: 258~265. doi: 10.1016/j.carbon.2018.09.060
-
[44]
C Zhao, L Liu, H Zhao et al. Nanoscale, 2014, 6(2): 882~888. doi: 10.1039/C3NR04532C
-
[45]
H Yan, M Cheng, B Zhong et al.Ionics, 2016, 22(11): 1999~2006. doi: 10.1007/s11581-016-1739-5
-
[46]
W Li, Q Zhang, G Zheng et al. Nano Lett., 2013, 13(11): 5534~5540. doi: 10.1021/nl403130h
-
[47]
H Tang, S Yao, J Mi et al. Mater. Lett., 2017, 186: 127~130. doi: 10.1016/j.matlet.2016.09.102
-
[48]
J Li, M Zhu, P Hu et al. Eur. J. Inorg. Chem., 2017, 2017(26): 3248~3252. doi: 10.1002/ejic.201700422
-
[49]
M S Song, S C Han, H S Kim et al. J. Electrochem. Soc., 2004, 151(6): A791~A795.
-
[50]
Y Zhang, Y Zhao, A Yermukhambetova et al. J. Mater. Chem. A, 2013, 1(2): 295~301. doi: 10.1039/C2TA00105E
-
[51]
R Yang, D Chen, B Ren et al. Mater. Lett., 2019, 235: 61~65. doi: 10.1016/j.matlet.2018.09.156
-
[52]
P Ji, T Zeng, X Hu et al. Solid State Ionics, 2018, 315: 52~58. doi: 10.1016/j.ssi.2017.12.004
-
[53]
K Shi, C Lai, X Liu et al. Energy Storage Mater., 2019, 17: 111~117. doi: 10.1016/j.ensm.2018.07.024
-
[54]
Y Wu, Q Xiao, S Huang et al. Mater. Chem. Phys., 2019, 221: 258~262. doi: 10.1016/j.matchemphys.2018.09.052
-
[55]
I A M Ousmane, R Li, C Wang et al. Micropor. Mesopor. Mater., 2018, 266: 276~282. doi: 10.1016/j.micromeso.2018.03.004
-
[56]
T Ma, M Liu, T Huang et al. J. Power Sources, 2018, 398: 75~82. doi: 10.1016/j.jpowsour.2018.07.048
-
[57]
Y Zhang, Y Zhao, A Yermukhambetova et al. J. Mater. Chem. A, 2013, 1: 295~301. doi: 10.1039/C2TA00105E
-
[58]
Y Zhang, Z Bakenov, Y Zhao et al. J. Power Sources, 2012, 208: 1~8. doi: 10.1016/j.jpowsour.2012.02.006
-
[59]
X Zhao, J K Kim, H J Ahn et al. Electrochim. Acta, 2013, 109: 145~152. doi: 10.1016/j.electacta.2013.07.067
-
[60]
X Liang, Y Liu, Z Wen et al. J. Power Sources, 2011, 196(16): 6951~6955. doi: 10.1016/j.jpowsour.2010.11.132
-
[61]
H J Noh, S T Myung, H G Jung et al. Adv. Funct. Mater., 2013, 23(8): 1028~1036. doi: 10.1002/adfm.201200699
-
[62]
Y Fu, A Manthiram. RSC Adv., 2012, 2(14): 5927~5929. doi: 10.1039/c2ra20393f
-
[63]
A Yermukhambetova, Z Bakenov, Y Zhang et al. J. Electroanal. Chem., 2016, 780: 407~415. doi: 10.1016/j.jelechem.2015.10.032
-
[64]
Q Liu, Q Jiang, L Jiang et al. Appl. Surf. Sci., 2018, 462: 393~398. doi: 10.1016/j.apsusc.2018.08.038
-
[65]
K Wu, B Shi, L Qi et al. Electrochim. Acta, 2018, 291: 24~30. doi: 10.1016/j.electacta.2018.09.086
-
[66]
D Zhang, Q Wang, Q Wang et al. Electrochim. Acta, 2015, 173: 476~482. doi: 10.1016/j.electacta.2015.05.086
-
[67]
X Gao, Y Shen, L Xing et al. Mater. Lett., 2016, 183: 413~416. doi: 10.1016/j.matlet.2016.07.059
-
[68]
D Xiao, C Lu, C Chen et al. Energy Storage Mater., 2018, 10: 216~222. doi: 10.1016/j.ensm.2017.05.015
-
[69]
X Li, L Zhang, Z Ding et al. J. Electroanal. Chem., 2017, 799: 617~624. doi: 10.1016/j.jelechem.2017.04.060
-
[1]
-

-

 下载:
下载:

 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 27
- 文章访问数: 2590
- HTML全文浏览量: 508
