引用本文:
梁坤, 张成华, 相宏伟, 杨勇, 李永旺. 改性SiO2对铁基催化剂H2、CO吸附及加氢性能的影响[J]. 燃料化学学报,
2019, 47(7): 769-779.
Citation:
LIANG Kun, ZHANG Cheng-hua, XIANG Hong-wei, YANG Yong, LI Yong-wang. Effects of modified SiO2 on H2 and CO adsorption and hydrogenation of iron-based catalysts[J]. Journal of Fuel Chemistry and Technology,
2019, 47(7): 769-779.
Received Date:
19 March 2019 Revised Date:
24 April 2019 Available Online:
10 July 2019
Fund Project:
The project was supported by the Major Research Program of National Natural Science Foundation of China (91545109)
Abstract:
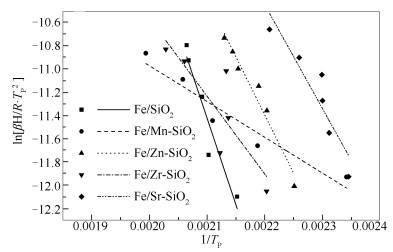
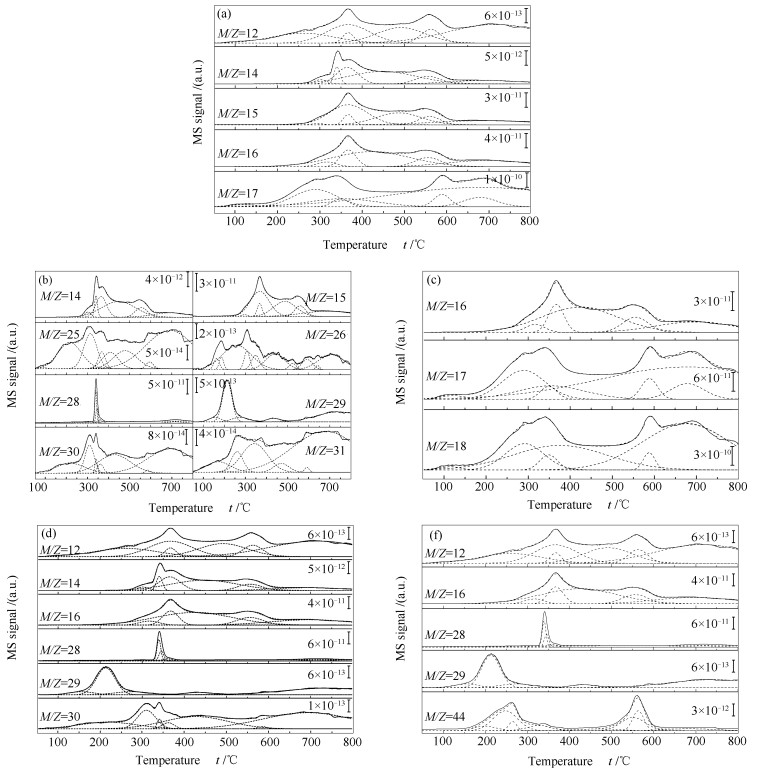
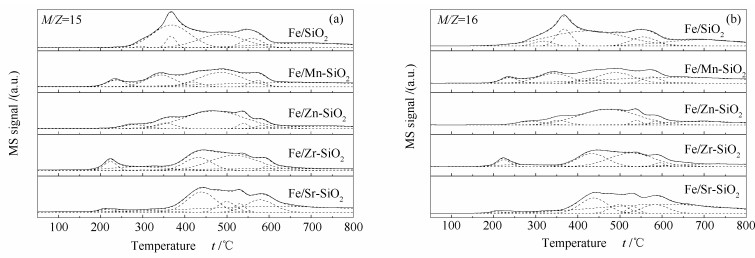
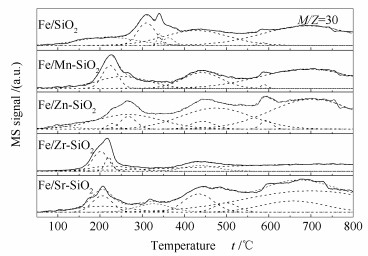
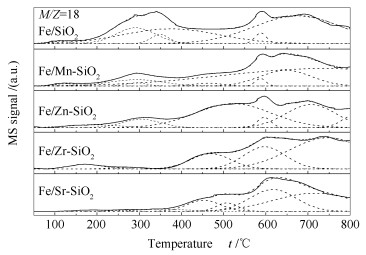
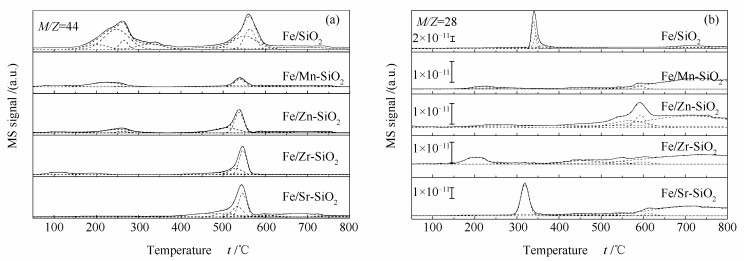
Metal-modified SiO2 supports were prepared by sol-gel method with Mn, Zn, Zr, and Sr cations doping, and then iron-based catalysts supported on modified SiO2 were prepared by the impregnation method. The iron-based catalysts were characterized by XRD, N2 adsorption, and XPS. The reduction adsorption property of H2 and hydrogenation property of CO were studied by temperature programmed methods. The interaction between catalyst and H was studied by kinetic analysis. The results indicate that metal modification has no influence on phase composition of Fe and surface electronic state of Fe species. However, the modification reduces surface area of catalyst and dispersion of active phase, weakens adsorption ability of H2 and lowers H2 desorption activation energy of the catalyst. Zn and Zr doping restrain reduction of catalysts, while Mn and Sr doping promotes reduction of catalysts. The doping of Mn, Zn and Zr inhibits adsorption of CO on the catalyst surface, while Sr promotes dissociation and adsorption of CO. Mn, Zn, Zr and Sr promote C-C coupling and hydrogenation reaction. Among them, Mn and Zr are more prominent.
DRY M E. The Fischer-Tropsch process:1950-2000[J]. Catal Today,
2002, 71(3):
227-241.
[2]
DRY M E. Present and future applications of the Fischer-Tropsch process[J]. Appl Catal A:Gen,
2004, 276(1):
1-3.
[3]
ZHANG Q H, KANG J C, WANG Y. Development of novel catalysts for Fischer-Tropsch synthesis:Tuning the product selectivity[J]. ChemCatChem,
2010, 2(9):
1030-1058.
doi: 10.1002/cctc.201000071
[4]
ZHANG Q H, DENG W P, WANG Y. Recent advances in understanding the key catalyst factors for Fischer-Tropsch synthesis[J]. J Energy Chem,
2013, 22(1):
27-38.
[5]
DE SMIT E, SWART I, CREEMER J F, HOVELING G H, GILLES M K, TYLISZCZAK T, KOOYMAN P J, ZANDBERGEN H W, MORIN C, WECKHUYSEN B M, DE GROOT F. Nanoscale chemical imaging of a working catalyst by scanning transmission X-ray microscopy[J]. Nature,
2008, 456(7219):
222-225.
doi: 10.1038/nature07516
[6]
CHANG Q, ZHANG C H, LIU C W, WEI Y X, AJIN V C, IULIAN DUGULAN A, NIEMANTSVERDRIET J W, LIU X W, He Y R, QING M, ZHENG L R, YUN Y F, YANG Y, LI Y W. Relationship between iron carbide phases (ε-Fe2C, Fe7C3, and x-Fe5C2) and catalytic performances of Fe/SiO2 Fischer-Tropsch catalysts[J]. ACS Catal,
2018, 8(4):
3304-3316.
doi: 10.1021/acscatal.7b04085
[7]
DE SMIT E, BEALE A M, NIKITENTO S, WECKHUYSEN B M. Local and long range order in promoted iron-based Fischer-Tropsch catalysts:A combined in situ X-ray absorption spectroscopy/wide angle X-ray scattering study[J]. J Catal,
2009, 262(2):
244-256.
[8]
DE SMIT E, CINQUINI F, BEALE A M. Stability and reactivity of ε-x-θ iron carbide catalyst phases in Fischer-Tropsch synthesis:Controlling μC[J]. J Am Chem Soc,
2010, 132(42):
14928-14941.
doi: 10.1021/ja105853q
[9]
SUO H Y, WANG S G, ZHANG C H, XU J, WU B S, YANG Y, XIANG H W, LI Y W. Chemical and structural effects of silica in iron-based Fischer-Tropsch synthesis catalysts[J]. J Catal,
2012, 286(3):
111-123.
[10]
陈嘉宁, 刘永梅. K、Mn助剂协同效应对Fe基催化剂上CO加氢制低碳烯烃反应性能的影响[J]. 燃料化学学报,
2013,41,(12): 1488-1494.
CHEN Jia-ning, LIU Yong-mei. Effects of Mn-K synergistic action on iron-based catalyst for CO hydrogenation to light olefins[J]. J Fuel Chem Technol,
2013, 41(12):
1488-1494.
[11]
TAO Z C, YANG Y, ZHANG C H, LI T Z, DING M Y, XIANG H W, LI Y W. Study of manganese promoter on a precipitated iron-based catalyst for Fischer-Tropsch synthesis[J]. J Nat Gas Chem,
2007, 16(3):
278-285.
doi: 10.1016/S1003-9953(07)60060-7
[12]
SAGLAM M. Effects of vanadium and zinc promotion on the olefin selectivity of iron Fischer-Tropsch catalysts[J]. Ind Eng Chem Res,
1989, 28(2):
150-154.
doi: 10.1021/ie00086a004
[13]
GAO X H, ZHANG J L, CHEN N, MA Q X, FAN S B, ZHAO T S, TSUBAKI N. Effects of zinc on Fe-based catalysts during the synthesis of light olefins from the Fischer-Tropsch process[J]. Chin J Catal,
2016, 37(4):
510-516.
doi: 10.1016/S1872-2067(15)61051-8
[14]
LI S Z, LI A W, KEISHNAMOORTHY S, IGLESIA E. Effects of Zn, Cu, and K promoters on the structure and on the reduction, carburization, and catalytic behavior of iron-based Fischer-Tropsch synthesis catalysts[J]. Catal Lett,
2001, 77(4):
197-205.
[15]
QING M, YANG Y, WU B, XU J, ZHANG C H, GAO P, LI Y W. Modification of Fe-SiO2 interaction with zirconia for iron-based Fischer-Tropsch catalysts[J]. J Catal,
2011, 279(1):
111-122.
[16]
LI J F, ZHANG C H, CHENG X F, QING M, XU J, WU B S, YANG Y, LI Y W. Effects of alkaline-earth metals on the structure, adsorption and catalytic behavior of iron-based Fischer-Tropsch synthesis catalysts[J]. Appl Catal A:Gen,
2013, 464/465(16):
10-19.
[17]
AMENOMIYA Y, CVETANOVIC R J. Application of flash-desorption method to catalyst studies. Ⅰ. Ethylene-alumina system[J]. J Phys Chem,
1963, 67(1):
2046-2049.
[18]
KOMERS R, AMENOMIYA Y, CVETANOVIC R J. Study of metal catalysts by temperature programmed desorption: Ⅰ. Chemisorption of ethylene on silica-supported platinum[J]. J Catal,
1969, 15(3):
293-300.
[19]
LIN H Q, QU H Y, CHEN W K, XU K, ZHENG J W, DUAN X P, ZHAI H S, YUAN Y Z. Promoted chemoselective crotonaldehyde hydrogenation on zirconia-doped SiO2 supported Ag catalysts:Interfacial catalysis over ternary Ag-ZrO2-SiO2 interfaces[J]. J Catal,
2019, 372(4):
19-32.
[20]
YAMASHITA T, HAYES P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials[J]. Appl Surf Sci,
2008, 254(8):
2441-2449.
doi: 10.1016/j.apsusc.2007.09.063
[21]
WACHS I E, DWYER D J, IGLESIA E. Characterization of Fe, Fe-Cu, and Fe-Ag Fischer-Tropsch catalysts[J]. Appl Catal,
1984, 12(2):
201-217.
[22]
XU J, BARTHOLOMEW C H, SUDWEEKS J, EGGET D L. Design, synthesis, and catalytic properties of silica-supported, Pt-promoted iron Fischer-Tropsch catalysts[J]. Top Catal,
2003, 26(1/4):
55-71.
[23]
ZHANG C H, YANG Y, TENG B T, LI T Z, XIANG H W, LI Y W. Study of an iron-manganese Fischer-Tropsch synthesis catalyst promoted with copper[J]. J Catal,
2006, 237(2):
405-415.
[24]
HARKNESS R W, EMMETT P H. Two types of activated adsorption of hydrogen on the surface of a promoted iron synthetic ammonia catalyst[J]. J Am Chem Soc,
1934, 56(2):
490-491.
[25]
WALCH S P. Model studies of the interaction of H atoms with BCC iron[J]. Surf Sci,
1984, 143(1):
188-203.
doi: 10.1016/0039-6028(84)90418-7
[26]
BLYHOLDER G, NEFF L D. Infrared study of the interaction of carbon monoxide and hydrogen on silica-supported iron[J]. J Phys Chem,
1962, 66(9):
1664-1667.
doi: 10.1021/j100815a024
[27]
WANG T, WANG S G, LUO Q Q, LI Y W, WANG J G, BELLER M, JIAO H J. Hydrogen adsorption structures and energetics on iron surfaces at high coverage[J]. J Phys Chem C,
2014, 118(8):
4181-4188.
doi: 10.1021/jp410635z
[28]
ROFER-DEPOORTER C K. A comprehensive mechanism for the Fischer-Tropsch synthesis[J]. Chem Rev,
1981, 81(5):
447-474.
doi: 10.1021/cr00045a002
[29]
MUETTERTIES E L, STEIN J. Mechanistic features of catalytic carbon monoxide hydrogenation reactions[J]. Chem Rev,
1979, 79(6):
479-490.
doi: 10.1021/cr60322a001

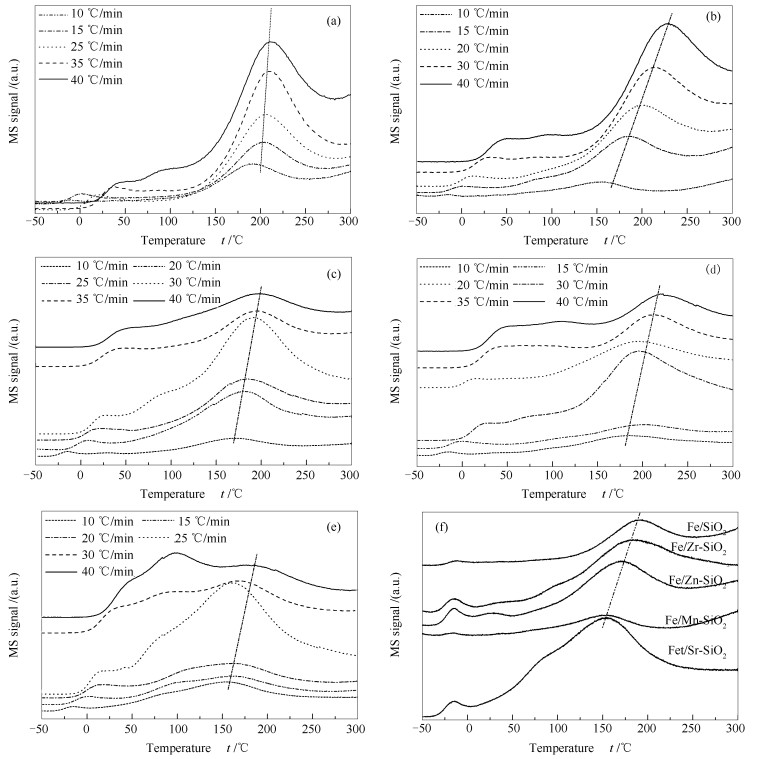
 下载:
下载:
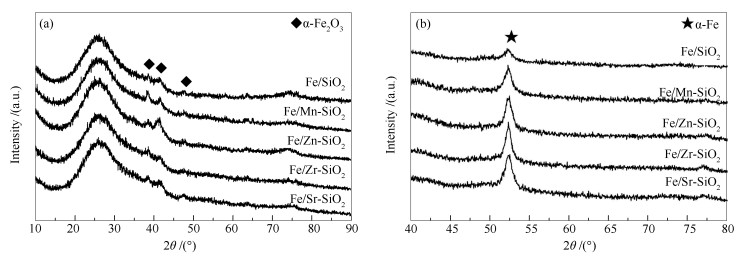
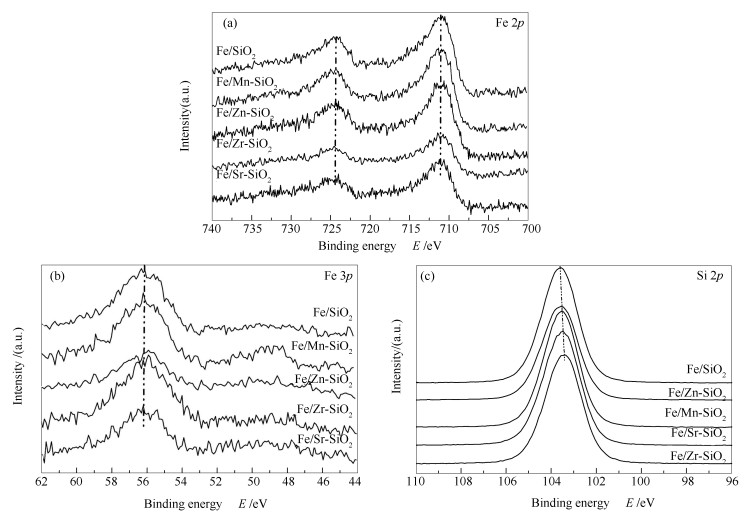
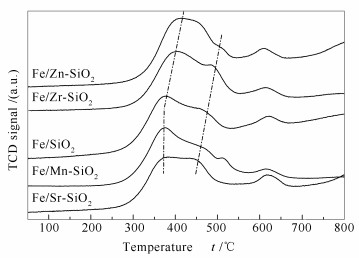

 下载:
下载:











