图式 1.
席夫碱大环L1及其还原产物L2的合成路线
Scheme 1.
Synthetic route of Schiff base macrocycle L1 and corresponding reduced macrocycle L2

由于阴离子识别在生命和环境领域的重要性, 使得新型阴离子受体的设计合成及其在主客体化学中的研究一直是活跃的研究领域[1, 2]。基于具有环状结构的有机分子能将环缘上的氢键供体(或受体)原子进行收敛排列, 使得大环与阴离子进行氢键作用的收敛能降低, 有利于提高大环主体与阴离子客体的络合作用能力, 系列含亚胺、酰胺等功能基团的有机大环分子相继被合成, 并在阴离子识别研究中发挥着积极作用[3~6]。在设计、合成含2, 6-二甲酰亚胺基吡啶单元的大环化合物及其阴离子识别研究方面, Sessler等[7, 8]设计合成了同时含2, 6-二甲酰胺基吡啶和双吡咯单元的环状化合物, 并在维持大环主体骨架结构基础上通过氧化、还原作用实现对环状骨架结构的刚、柔性调整, 达到对系列阴离子氢键识别作用的对比考察。结果表明, 大环对四面体构型的阴离子(硫酸根或磷酸根)的氢键作用能力比对球形卤阴离子或平面三角形构型的NO3-、CH3 COO-等具有更好的选择性识别能力, 同时还表明, 环状骨架结构的刚、柔性的微小变化, 对大环主体与阴离子的选择性识别性质有着重要影响。在关于生物体系模拟研究方面, 针对磷酸根(Pi)识别与探针研究一直是研究者广泛重视的课题[9, 10], 相比之下, 含两个P-O四面体中心的焦磷酸根(PPi)比Pi具有更大的体积和更多的负电性, 在生物体能量储存及信号传输方面具有更多的生理相关性, 但在设计合成相应的大环配体用于PPi和Pi的识别的对比考察方面的研究却相对较稀缺。近年来, 笔者课题组在合成系列含2, 6-二甲酰胺基吡啶单元的大环化合物并开展其与阴离子识别或非共价作用超分子组装研究方面取得了一些进展[11~14]。本文通过N, N′-(3-氨基苯基) -2, 6-二甲酰亚胺吡啶(1)和5, 5′-亚甲基双水杨醛(2)进行缩合反应得到含酚羟基的[1 + 1]席夫碱大环L1, 在此基础上将L1中席夫碱C = N双键还原得到环状骨架更具柔性的饱和大环L2(图式 1), 进一步将其用于对系列阴离子进行络合作用的对比考察。
Ry-2型熔点仪(温度计未校正); JEOL ECX 400 MHz核磁共振谱仪; Vario EL III型元素分析仪; 岛津UV-2700紫外分光光度计; 安捷伦6500质谱仪; Bruker Smart Apex X射线单晶衍射仪。所用试剂和药品均为市售分析纯级。
前体二胺(1)的合成:按照文献[15]所报道的方法合成, 为乳白色固体, 产率63%, 熔点281 ~ 283℃;1H NMR (400MHz, DMSO-d6) δ:10. 76 (s, 2H, -NH), 8. 2 ~ 8. 36 (m, 3H, py-H), 6. 36 ~ 7. 18(m, 8H, Ar-H), 5. 18(s, 4H, -NH2)。
前体二醛(2)的合成:按照文献[16]的方法合成, 为淡黄色固体, 产率49. 0%, 熔点139 ~ 142℃;1 H NMR (400MHz, CDCl3) δ: 10. 02 (s, 2H, -CHO), 7. 65 (s, 2H, Ar-H), 7. 48 (d, 2H, ArH), 6. 93(d, 2H, Ar-H), 3. 99(s, 2H, -CH2-)。
称取0. 078g (0. 225mmol)化合物1置于250mL三口瓶中, 加入100mL无水甲醇, 加热回流至溶解, 搅拌下向上述溶液中加入0. 058g (0. 225mmol)化合物2的20mL无水甲醇溶液, 反应一段时间后溶液逐渐变浑浊, 并且有橘黄色固体析出, 回流24h后停止反应, 过滤, 滤饼用热甲醇洗涤3次, 干燥后得橘黄色固体0. 083g, 产率65. 0%, 熔点> 300℃。1H NMR (400MHz, DMSO-d6) δ: 11. 99 (s, 2H, -OH), 11. 18 (s, 2H, -CONH-), 9. 00 (s, 2H, -CH = N-), 8. 43 (t, J = 7. 6Hz, 2H, Py-H), 8. 43(t, J = 7. 6Hz, 2H, Ar-H), 8. 31 (t, J = 8. 0Hz, 1H, Py-H), 7. 88 (s, 2H, Ar-H), 7. 54(s, 2H, Ar-H), 7. 48(t, J = 8. 0Hz, 2H, Ar-H), 7. 34(d, J = 8. 4Hz, 2H, Ar-H), 7. 18(d, J = 8. 0Hz, 2H, Ar-H), 6. 85(d, J = 8. 4Hz, 2H, Ar-H), 3. 84 (s, 2H, -CH2-);13C NMR (101MHz, DMSOd6) δ:162. 5, 162. 2, 158. 4, 149. 8, 149. 3, 140. 6, 139. 8, 134. 0, 133. 4, 131. 6, 130. 5, 126. 3, 120. 5, 119. 3, 119. 0, 117. 0, 112. 5, 40. 9; IR (KBr, ν/cm-1) : 3413, 1619, 1537; FABMS m/z: 566. 18 [M-H]+; 元素分析, C34 H25 N5 O4 :理论值: C 71. 95, H 4. 44, N 12. 34;实测值: C 71. 83, H 4. 57, N 12. 43。
取L1溶解于DMF和乙腈的混合溶液中, 常温下培养, 7d后长出适合X-射线单晶衍射测定的橘黄色针状晶体。
在含有40mL四氢呋喃的三口瓶中加入0. 50g (0. 88mmol) L1, 在25℃下搅拌, 将0. 15g (4. 0mmol) NaBH4分3次加入(每间隔10min加0. 05g)到上述溶液中, 室温下反应8h, 然后旋蒸除去四氢呋喃, 加入50mL蒸馏水, 静置, 过滤, 真空干燥得淡黄色固体L2, 经400目硅胶柱层析, 以石油醚/乙酸乙酯(体积比1 :2)为淋洗剂, 收集第一带, 减压旋蒸除去溶剂, 得乳白色固体0. 27g, 产率为54%, 熔点> 300℃。1H NMR (400MHz, DMSO-d6) δ: 10. 91 (s, 2H, -CONH-), 9. 33(s, 2H, -OH), 8. 29(m, 2H, Py-H), 8. 29 (m, 1H, Py-H), 7. 67 (d, J = 7. 6Hz, 2H, Ar-H), 7. 24 (s, 2H, Ar-H), 7. 12(t, J = 8. 0Hz, 2H, Ar-H), 6. 97 (d, J = 8. 4Hz, 2H, Ar-H), 6. 94 (s, 2H, Ar-H), 6. 72(d, J = 8. 4Hz, 2H, Ar-H), 6. 51(d, J = 8. 0Hz, 2H, Ar-H), 5. 55 (t, J = 9. 6Hz, 2H, Ar-NH-), 4. 15 (d, J = 5. 2Hz, 4H, -CH2 -), 3. 65 (s, 2H, -CH 2 -);13 C NMR (101MHz, DMSO-6) δ:161. 7, 153. 5, 150. 0, 149. 4, 140. 7, 139. 2, 133. 2, 130. 1, 129. 7, 128. 4, 125. 4, 125. 2, 115. 3, 108. 6, 108. 4, 106. 4, 44. 9, 42. 4;IR (KBr, ν/cm-1) :3415, 1615, 1558;FABMS m/z: 570. 21 [M-H]+; 元素分析, C34H29N5O4:理论值:C 71. 44, H 5. 11, N 12. 25;实测值:C 71. 31, H 5. 19, N 12. 36。
取L2溶解于DMSO和乙腈的混合溶液中, 常温下培养, 7d后长出适合X-射线单晶衍射测定的白色晶体。
选取大小适当的晶体, 用Bruker Smart Apex单晶衍射仪采用经石墨单色器单色化的Mo Kα射线(λ = 0. 071073nm)以φ-ω扫描方式收集单晶衍射数据。强度数据进行了经验吸收校正、LP校正。晶体结构由直接法解得, 对全部非氢原子坐标及其各向异性热参数进行了全矩阵最小二乘法修正, 所有计算用SHELX-97程序完成[17]。
L1母液的配制:以DMSO /乙腈(1 : 99, 体积比, 下同)为溶剂, 将L1配成浓度为1 × 10-4mol· L-1的溶液, 移取20. 0mL于100mL的容量瓶中, 稀释成浓度为2×10-5 mol· L-1的溶液, 用DMSO和乙腈的混合溶剂定容, 再取3mL上述溶液于9个5mL的塑料离心管中, 分别加入0. 15mL浓度均为2×10-3 mol· L-1的系列阴离子(F-, Cl-, Br-, I-, NO3-, AcO-, H2PO4-, HP2O73-, H2P2O72-)的季铵盐(TBA+ X-)的乙腈溶液, 混匀、放置达体系吸光度稳定不变后, 室温下在200 ~ 600 nm范围内记录各反应体系的紫外-可见吸收光谱(DMSO /乙腈(1 : 99)作参比)。
室温下, 移取浓度为2. 00 × 10-5mol·L-1的L1的DMSO和乙腈混合溶剂溶液, 分别每次滴加浓度为2. 00×10-3mol· L-1、体积不同的H2PO4-离子的(TBA+ X-)盐的乙腈溶液, 得到阴离子浓度依次增加的系列反应体系([H2PO4-]/[L1]的物质的量比在0. 1 ~ 5. 0)。同样, 按照上述方法配制HP2O73-和H2P2O72-离子的乙腈溶液。待反应达平衡后, 于200 ~ 600 nm范围内测定其UV-Vis光谱, 并通过摩尔比法确定配位反应的配位比。
进一步采用Job's法对主客体配位比进行确定, 即以DMSO和乙腈(1 : 99)混合溶液为溶剂, 分别配制浓度均为1. 00 × 10-4mol· L-1的L1与离子H2PO4-、HP2O73-和H2P2O72-的操作液。固定各配位反应体系中反应物总浓度为4. 00×10-5mol· L-1, 配制L1与H2PO4-、HP2O73-和H2P2O72-离子浓度比不同的反应体系。待系列体系达反应平衡后, 测定体系的吸光度。
大环L1的1H NMR谱中, 两个酚羟基质子的δ在11. 99处(单峰), 酰亚胺(CONH)质子的δ在11. 18处(单峰), 两个席夫碱(CH = N)质子的δ在9. 00处(单峰), 吡啶环上处在取代基邻位的2个质子的δ在8. 43处(三重峰), 处在取代基间位的质子的δ在8. 31处(三重峰)。在经由酰亚胺基与吡啶环相连的两个苯环上, 位于席夫碱间隔基对位的2个质子的δ在8. 43处(三重峰), 处于两个取代基之间的2个质子的δ在7. 54处(单峰), 处于取代基间位的2个质子的δ在7. 48处(三重峰), 处于酰亚胺取代基对位的2个质子的δ在6. 85处(双重峰)。在亚甲基连接的两个苯环上, 处在亚甲基和席夫碱间隔基之间的两个质子的δ在7. 88处(单峰), 处于席夫碱间隔基对位的质子的δ在7. 33处(双重峰), 处于酚羟基邻位的质子的δ在7. 17处(双重峰), 两个亚甲基质子的δ在3. 84处, 呈现为单峰。L2的1H NMR谱中, 与席夫碱大环L1相比, 相应的质子峰进一步向高场发生位移, 这源于大环L1被还原后, 分子骨架结构更具柔性, 能量进一步增加。其中, 两个甲酰亚胺(CONH)质子的δ出现在10. 91处, 两个酚羟基质子的δ在9. 33处。与L1相比, 在L2的1H NMR谱中2个CH = N质子峰消失, 经还原得到的结构单元(-CH2 -NH-)中的2个NH质子的δ出现在5. 55处, 2个亚甲基质子的δ出现在4. 15处。
L1和L2的质谱(FABMS)数据表明(见附件中S2), L1的分子离子峰[M-H]+ m/z为566. 18, L2的分子离子峰[M-H]+ m/z为570. 21, 表明前体二胺(1)和二醛(2)按1 : 1的摩尔比缩合形成了[1+1]的席夫碱大环L1, L2为L1中两个席夫碱C = N双键被还原的产物。谱学表征结果与这两个大环的单晶结构相吻合。
通过单晶培养和结构解析, 获得了L1和L2的晶体结构数据, 存于英国剑桥数据中心, CCDC: 1975240, 1975241。
[1+1]席夫碱大环L1的分子结构如图 1。在该大环分子中, N(4) ― C(14)和N(5) ― C(28)的键长分别为0. 1280(4) nm和0. 1273(4) nm, 接近于典型的碳氮双键键长(平均键长为0. 126nm[18]), 通过酰亚胺基与中央吡啶连接的两个苯环之间的二面角为157. 1°, 以亚甲基连接的两个酚环之间的二面角为124. 01°, 因酚羟基O(4)、O(3)分别与其相邻的N(4)、N(5)形成O ― H… N氢键作用, 所形成的五元环分别与相邻的酚环共面, 进一步增强了席夫碱大环的骨架刚性, 大环骨架呈现为非平面结构。在相应的还原大环L2结构中, 因两个席夫碱C = N双键被还原, 使得大环柔性进一步增加, 亚甲基连接的两个酚环上酚羟基氧原子中O(3)与相邻的亚胺氮N (4)形成O ― H… N氢键作用, 而另一个酚羟基氧原子中O (4)朝向环外, 导致亚甲基连接的两个酚环之间的二面角为112. 4°, 与席夫碱大环L1相比, 大环L2呈环状骨架进一步扭曲的非平面结构。
向浓度均为2. 00 × 10- 5mol· L的L1或L2的DMSO /乙腈(1 : 99)混合溶液中, 分别滴加5. 00倍量的系列阴离子(即F-, Cl-, Br-, I-, NO3-, AcO-, H2 PO4-, HP2O73-, H2P2O72-)的季铵盐(TBA+ X-), 室温下在200 ~ 600 nm范围内记录各反应平衡体系的UV-Vis光谱变化(图 2)。从图 2(a)可见, 大环L1仅能与P-O键呈四面体构型的H2PO4-、HP2O73-、H2P2O72-表现出明显的络合作用, 而L2与所考察的系列阴离子的作用均不明显(图 2b)。据此, 进一步考察了L1分别与上述3种阴离子的氢键作用。
向浓度均为2. 00 × 10- 5mol· L- 1的L1的DMSO /乙腈(1 :99)混合溶剂溶液中, 分别滴加浓度为2. 00 × 10- 3 mol · L- 1的H2PO4-、HP2O73-、H2P2O72-的TBA盐(0. 1 ~ 5. 0 eq), UV-Vis光谱滴定曲线如图 3所示。从图 3(a)可见, 随着H2PO4-离子滴加量的不断增加, L1在272nm处的最大吸收强度逐渐减弱, 最大吸收峰逐渐红移到274nm, 并在280nm处出现一等吸收点。选用选择性较高的308nm处不同H2 PO4-浓度时作用体系吸光度A对H2 PO4-与L1的物质量比n (H2 PO4-)/n(L1)作图(见插图), 可见当L1与H2 PO4-物质的量比为1 : 1时, 体系吸光度值出现转折并趋向平缓, 表明溶液中L1与H2 PO4-形成了络合比为1 :1的配合物。类似地, 依次获得L1分别与HP2O73-或H2P2O72-作用体系的UV-Vis光谱滴定曲线(图 3(b) 和图 3((c))。由图可见, 随着HP2O73-或H2P2O72-加入量的不断增加, L1与阴离子作用过程中, 在272nm处的最大吸收均逐渐减弱, 等吸收点分别出现在278、276nm处。分别选用L1与HP2O73-或H2P2O72-作用过程中选择性较高波长(λmax分别为309、339nm)处体系的吸光度A对大环L1与H2P2O72-的物质量比n(HP2O73-)/n(L1)或n (H2P2O72-)/n (L1)作图(见插图), 结果表明, L1与HP2O73-或H2P2O72-作用均形成络合比为1 :1的配合物。
进一步采用Job’s法确定反应的络合比。将L1分别与不同浓度的H2PO4-作用体系的ΔA对相应的浓度比[H2PO4-]/([L1] +[H2PO4-])作图(图 4(a)), 表明L1与H2PO4-形成1 :1的配合物。类似地, 将L1分别与阴离子HP2O73-或H2P2O72-作用的各识别体系的ΔA对相应的浓度比[HP2O73-]/([L1] + [HP2O73-]或[H2P2O72-]/([L1] +[H2P2O72-])作图(图 4(b)和(4(c)), 表明L1分别与HP2O73-或H2P2O72-作用也均形成1 :1的配合物, Job's法获得结果与摩尔比法的一致。
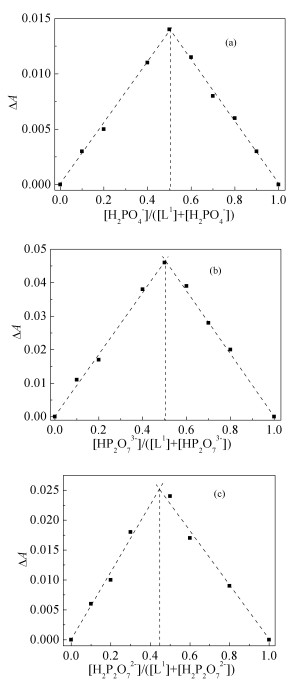
(a) H2PO4-, (b)HP2O73-, (c) H2P2O72-
平衡常数的测定:基于L1与H2PO4-、HP2O73-、H2P2O72-络合比均为1 :1, 参照文献[19]中所提供的方法, 按照式(1)计算得到反应平衡常数K。
|
$ \begin{array}{l} \Delta {A_{obs}} = {\varepsilon _{\Delta HG}}\frac{1}{2}\left( {{G_0} + {\rm{ }}{H_0} + \frac{1}{{{K_a}}}} \right) - \\ \sqrt {{{\left( {{{\left[ G \right]}_0} + {{\left[ H \right]}_0} + \frac{1}{{{K_a}}}} \right)}^2} + 4\left[ {{H_0}} \right]\left[ {{G_0}} \right]} \end{array} $ |
(1) |
式中, ΔAobs是在阴离子(客体)存在条件下配合物的吸光度与自由主体吸光度之差; [G]0和[H]0分别是客体(阴离子)和主体(大环)的初始浓度; εΔHG是配合物HG与自由主体的摩尔吸光系数之差; Ka为主体大环分子和客体阴离子的结合常数。经拟合得席夫碱大环L1与H2PO4-的结合常数Ka = 1. 52 × 105 L· mol- 1, 与HP2O73-或H2P2O72-的Ka分别为8. 80 × 105和3. 62 × 106 L· mol-1, 络合能力按照H2PO4-、HP2O73-、H2P2O72-顺序依次增加。
为进一步阐释L1分别与H2PO4-、HP2O73-或H2P2O72-发生氢键作用的络合位点, 在DMSO-d6中采用1H NMR滴定技术对反应过程中大环L1的氢谱信号变化情况进行考察(图 5)。
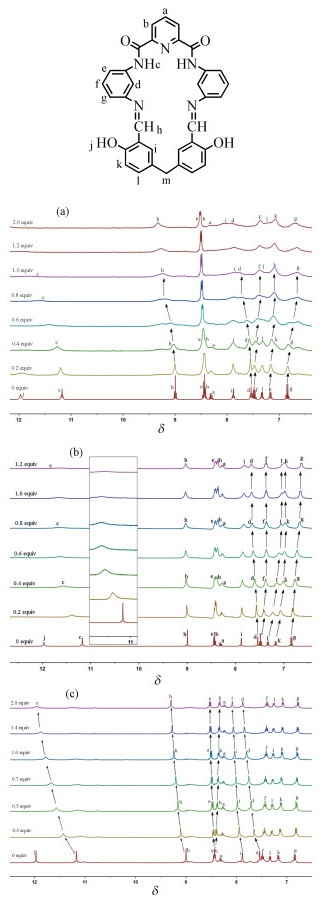
(a) H2PO4-, (b)HP2O73-, (c) H2P2O72-
从图 5(a) 可见, 随着H2PO4-的不断增加。酰亚胺质子(Hc)、席夫碱CH = N质子(Hh)和苯环质子(Hd)均向低场发生不同程度的位移, 峰形变宽且强度逐渐减弱, 当加入1倍量H2PO4-时, 相应3个质子向低场位移的Δδ依次为0. 62、0. 27和0. 20。同时, 因大环与H2PO4-产生氢键作用导致苯环上的质子(Hf, Hk, Hg)向高场发生不同程度的位移。在图 5(b)中, 随着主客体作用体系中HP2O73-浓度不断增加, L1中仅酰亚胺质子(Hc)和苯环上的质子(Hd)的δ不断往低场发生不同程度位移且蜂形变宽, Δδ分别为0. 47和0. 12, 但席夫碱CH = N质子(Hh)的δ无明显变化, 表明大环中酰亚胺质子(Hc)和苯环上的质子(Hd)与HP2O73-发生了氢键作用。相比之下, L1与H2P2 O72-作用体系中, 随着H2P2O72-的不断滴加, 酰亚胺质子(Hc)、席夫碱CH=N质子(Hh)、苯环质子(Hd)和亚甲基连接的苯环质子(Hi)的δ均不断向低场位移, Δδ依次为0. 76、0. 30、0. 20和0. 34, 表明席夫碱大环L1中这些质子都与H2P2O72-发生了氢键作用。
综上所述, 从大环L1晶体结构解析结果可见, 在L1分子环状骨架上2个酚羟基的取向均指向环的外侧, 并与相邻席夫碱氮原子发生氢键作用, 立足与主客体氢键作用的空间因素考虑, 可以推测这2个酚羟基与外加阴离子之间将不易发生氢键作用。依据UV-Vis光谱滴定获得的稳定常数结果, 即席夫碱大环L1与仅有1个四面体氢键作用单元的H2PO4-进行氢键作用的能力比具有两个四面体氢键作用单元的HP2O73-和H2P2O72-均弱,源于具有两个氢络合作用单元的焦磷酸根与大环作用在形成氢键作用的数目或进行氢键作用的空间取向方面更加有利。
Molina P, Zapata F, Caballero A. Chem. Rev., 2017, 117(15): 9907~9972. doi: 10.1021/acs.chemrev.6b00814
He Q, Kelliher M, Bähring S, et al. J. Am. Chem. Soc., 2017, 139(21): 7140~7143. doi: 10.1021/jacs.7b02329
Zhou H J, Zhao Y S, Gao G, et al. J. Am. Chem. Soc., 2013, 135(40): 14908~14911. doi: 10.1021/ja406638b
Chandra B, Mahanta S P, Pati N N. Org. Lett., 2013, 15(2): 306~309. doi: 10.1021/ol3032158
Ghite W N, Lovett H G, Beer P D. RSC Adv., 2014, 4: 12133~12147. doi: 10.1039/C4RA00615A
Izawa H, Nishino S, Sumita M. Chem. Commun., 2015, 51(41): 8596~8599. doi: 10.1039/C5CC01709B
Sessler J L, Katayev E, Pantos G D et al. J. Am. Chem. Soc., 2005, 127(32): 11442~11446. doi: 10.1021/ja0522938
陈冬梅, 吴娟, 欧敏, 等.化学通报, 2015, 78, (5): 447~453. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20140917003&flag=1
Ravikumar I, Ghosh P. Chem. Soc. Rev., 2012, 41(8): 3077~3098. doi: 10.1039/c2cs15293b
Dey S K, Das G. Dalton Transac., 2012, 41(29): 8960~8972. doi: 10.1039/c2dt30687e
张文龙, 黄超, 刘兴丽, 等.有机化学, 2017, 37(2): 474~479.
刘兴丽, 汤正河, 朱必学, 等.有机化学, 2017, 37(11): 2911~2918.
Chen H Y, Huang C, Ding Y Z, et al. Chem. Sci., 2019, 10: 490~496. doi: 10.1039/C8SC03824D
Chen H Y, Huang C, Deng Y X, et al. ACS Nano., 2019, 13: 2840~2848. doi: 10.1021/acsnano.8b09478
张奇龙, 徐红, 席晓岚.分子科学学报, 2011, 27(6): 402~406.
Marvel C S, Tark N. J. Am. Chem. Soc., 1957, 79(22): 6000~6002. doi: 10.1021/ja01579a041
Sheldrick G M. SHELX-97, University of Göttingen, Germany, 1997.
zkar S, Ülkü D, Yildirim L T, et al. J. Mol. Struct., 2004, 688(1/3): 207~211.
Thordarson P. Chem. Soc. Rev., 2011, 40(3): 1305~1323. doi: 10.1039/C0CS00062K

图式 1 席夫碱大环L1及其还原产物L2的合成路线
Scheme 1 Synthetic route of Schiff base macrocycle L1 and corresponding reduced macrocycle L2

图 1 L1(a)和L2(b)的分子结构(椭球几率为30%)
Figure 1 Molecular structures of L1(a) and L2(b) (probability of ellipsoid is 30%)

图 2 向大环的DMSO /乙腈(1 :99)溶液中分别加入5. 00倍量阴离子(Xn-)的UV-Vis光谱
Figure 2 UV-Vis absorption spectra of the macrocycles in DMSO/CH3CN (V/V = 1 :99) solution upon addition of 5. 0 equiv. anion Xn-. Xn- =F-, Cl-, Br-, I-, NO3-, AcO-, H2PO4-, HP2O73-, H2P2O72-

图 3 大环L1的吸收光谱随H2 PO4-(a)、HP2O73-(b)、H2P2O72-(c)的滴加量的变化曲线
Figure 3 Variation curve of the absorption spectra of macrocyclic L1 with the amount of H2 PO4-4(a), HP2O73-(b), and H2P2O72-(c)

图 4 L1分别与阴离子作用体系的Job's图
Figure 4 Job' s plots of L1 with anions
(a) H2PO4-, (b)HP2O73-, (c) H2P2O72-

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
