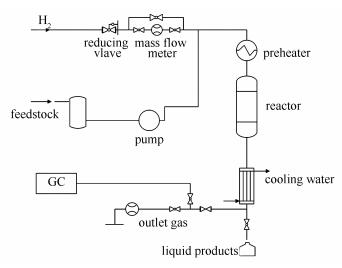
 图1
生物油催化加氢固定床反应系统示意图
Figure1.
Fixed-bed reaction system for bio-oil catalytic hydrogenation
图1
生物油催化加氢固定床反应系统示意图
Figure1.
Fixed-bed reaction system for bio-oil catalytic hydrogenation
引用本文:
陈军昊, 卢亮, 王树荣. 基于分子蒸馏的模拟生物油温和加氢研究[J]. 燃料化学学报,
2017, 45(9): 1056-1063.

Citation: CHEN Jun-hao, LU Liang, WANG Shu-rong. Mild hydrogenation of simulated bio-oil based on molecular distillation[J]. Journal of Fuel Chemistry and Technology, 2017, 45(9): 1056-1063.
Citation: CHEN Jun-hao, LU Liang, WANG Shu-rong. Mild hydrogenation of simulated bio-oil based on molecular distillation[J]. Journal of Fuel Chemistry and Technology, 2017, 45(9): 1056-1063.
基于分子蒸馏的模拟生物油温和加氢研究
English
Mild hydrogenation of simulated bio-oil based on molecular distillation
Abstract:
A mild hydrogenation of simulated bio-oil was carried out in a fixed-bed reactor. Based on the experimental results, 300℃/4 MPa was chosen as an optimum condition for the mild hydrogenation, the simulated bio-oil was nearly completely converted. Besides, the liquid product selectivity achieved 85.0% and its (H/C)eff was significantly promoted from 1.266 to 1.554. The liquid composition was greatly improved and a notable decrease of phenols and acids contents was observed. In this case, the product activity was significantly enhanced and then the subsequent catalytic cracking was favored.
-
Key words:
- bio-oil
- / model compounds
- / mild hydrogenation
- / effective hydrogen to carbon ratio
-
生物油可以通过生物质快速热裂解制取得到,被认为是传统运输燃料的替代品。然而,粗制生物油的高含氧量、高含水量、腐蚀性和低热值等问题一直限制了生物油的高品位利用[1, 2]。因此,须对生物油进行提质改性。
目前,生物油提质改性技术主要包括催化裂化、催化加氢、酯化、重整制氢以及乳化等[3-5]。其中,催化裂化被认为是很有前景的生物油改性技术,通过将生物油中的氧以CO、CO2和H2O的形式脱除,从而将生物油转化为富含芳香烃的液体燃料[6]。然而,粗制生物油成分十分复杂,由酮、酸、醛、酚和糖类等组成,反应活性不一。关于生物油模化物催化裂化的一系列研究表明,酮类和酸类具有向烃类转化的潜力,而大分子酚类聚合物和糖类则明显倾向于生成焦炭,从而导致催化剂的快速失活[7-9]。因此,生物油中酚类聚合物和糖类的脱除对于提高其反应活性有重要意义。分子蒸馏是一种高效的分离技术,可以将生物油划分为反应活性较高的蒸出馏分和难以转化的残余馏分,其中,蒸出馏分富集了主要的酮类和酸类,而裂化活性较差的酚类聚合物和糖类则基本保留在残余馏分[10, 11]。因此,基于生物油蒸出馏分的催化裂化研究具有更加显著的应用前景。虽然蒸出馏分中主要含有的酸类和酮类反应活性较高,但其仍然具有很高的含氧量和不饱和度,从而表现出一定的积炭趋势。为了反映生物油各组分的含氧量与不饱和度对其裂化反应活性的影响,研究者引入了有效氢碳比[(H/C)eff]的概念[12],如公式(1) 所示,H、C、O分别代表对应化合物中三种元素的质量分数。
Mentzel等[12]在生物油模化物的裂化研究中发现,较高的有效氢碳比对于提高裂化过程催化剂的稳定性有积极作用。典型生物油的有效氢碳比小于1.0,而主要目标产物单环芳烃的有效氢碳比为1.0-2.0,因此, 必须对反应原料进行处理以提高其有效氢碳比,从而有效促进目标产物生成并提高反应稳定性。Wang等[13-16]引入乙醇作为共裂化反应物开展了生物油蒸出馏分及模化物的系列研究,获得了较高的油相产率,同时焦炭产率明显降低。
催化加氢是另一种常用的生物油改性技术,主要包括两种形式:一种是加氢脱氧,即在高压下使生物油中的氧以H2O的形式脱除,将生物油转化为脂肪烃类产物[17-19],这种方式为了保证足够的脱氧效率,实验中的操作压力为10-20MPa甚至更高,氢气消耗量较大,同时对设备的要求也更高;另一种是温和加氢,即在较低的压力下使生物油中的不饱和双键饱和,从而提高生物油的稳定性[20-25]。典型的加氢催化剂包括贵金属[20, 22-24]、钼基硫化物[26, 27]、金属磷化物[28]以及其他金属催化剂[21, 25],其中,Pd基催化剂在研究中表现出很高的加氢活性[20, 23]。鉴于生物油有效氢碳比很低,如果能对其先进行温和加氢处理,然后再用于催化裂化,就可以进一步改进生物油提质改性工艺。Vispute等[29]将催化裂化和温和加氢相结合,先通过固定床反应器在10MPa条件下对生物油水相进行催化加氢,再将反应后冷凝收集到的产物二次汽化送入流化床反应器进行催化裂化,得到的焦炭选择性由单独裂化的32.3%明显降低至12.6%。为了探索合适的加氢工况以改进生物油的整体裂化改性工艺,在本研究中,作者开展温和条件下(2-4MPa、200-300℃)的模拟生物油催化加氢研究,在3%Pd/nano-SiO2的催化作用下,首先, 使反应物中的不饱和双键饱和并转化为对应的醇类,显著提高生物油的整体有效氢碳比,从而抑制后续裂化过程中的积炭,提高裂化过程的活性和稳定性。研究选取生物油蒸出馏分中的典型模化物,包括羟基丙酮、环戊酮、乙酸、愈创木酚、苯酚和糠醛,另外乙醇作为溶剂。
1 实验部分
1.1 化学药品
Nano-SiO2和PdCl2从阿拉丁试剂公司购买。加氢反应使用的催化剂Pd/nano-SiO2通过浸渍法制备,Pd的负载量为3%(质量分数)。在室温下将一定量的PdCl2加入去离子水中并用盐酸溶解直至pH值为3,然后加入焙烧过的nano-SiO2并静置12h,再将混合物在110℃烘箱中烘干过夜,最后将样品在550℃下焙烧6h,并压片筛分至40-60目。
实验中选取的模拟生物油组成包括羟基丙酮(Alfa Aesar)、环戊酮(Aladdin)、乙酸(中国国药集团)、愈创木酚(Alfa Aesar)、苯酚(中国国药集团)和糠醛(中国国药集团),各模化物的配比依据其在生物油蒸出馏分中的分布[13],模拟生物油与乙醇的比例为1:1。最终反应原料的组成为20%羟基丙酮、5%环戊酮、15%乙酸、5%愈创木酚、2.5%苯酚、2.5%糠醛和50%乙醇。
1.2 催化剂的活性测试
催化实验在图 1所示的固定床反应系统上进行,反应器为内径8mm的不锈钢管,催化剂装填量为3g,催化剂上下两端均使用石英棉支撑,其中, Pd/nano-SiO2催化剂预先在30mL/min的氢气气流和350℃的温度下进行2h的还原处理。反应物通过HPLC泵给料,经过雾化后与H2一起进入反应器中。反应压力由H2维持,其流量为30mL/min。反应物的质量空速(WHSV)为1h-1。反应器出口的气体经过冷凝器进行冷却,气水分离后得到液体产物和不可冷凝的气体。实验中加氢温度为200-300℃,氢气压力为2-4MPa。
图1
生物油催化加氢固定床反应系统示意图
Figure1.
Fixed-bed reaction system for bio-oil catalytic hydrogenation
实验获得的气体产物通过在线气相色谱(华爱9560) 进行定量分析。柱箱在50℃下保持3min,然后以5℃/min的速率升高到120℃,并保持13min。温和加氢的液体产物为均一相,通过TraceDSQⅡ型气质联用仪(GC-MS)确定化合物的结构,并采用面积归一法计算各物质的相对含量,采用的色谱柱为Agilent公司生产的DB-WAX极性色谱柱,柱箱在40℃下保持1min,然后以8℃/min的速率升高到240℃,并保持10min。剩余的液体反应物通过气相色谱仪(Agilent 7890A)利用外标法进行定量,以计算反应物的转化率,所用色谱柱为INNOWAX capillary column,升温程序与GC-MS一致。反应得到的液体产物通过Vario Micro元素分析仪对C、H、O进行分析,计算液体产物的有效氢碳比。单个反应物的转化率(xi, i=羟基丙酮、环戊酮、乙酸、苯酚、愈创木酚和糠醛),模拟生物油的整体转化率x和产物选择性(sj,j=液体产物和各种气体产物)通过公式(2)-公式(4) 进行定义。在选择性的计算中,未转化的反应物不计入液体产物中。以下公式中的符号“m”和“w”分别代表相应物质的质量和质量分数。
Pd粒子的分散性及颗粒大小采用Philips-FET公司TecnaiG2F30型高分辨率透射电子显微镜进行检测。反应后催化剂的焦炭含量通过热重进行分析,使用TGA/SDTA851e热重分析仪。样品在30mL/min的空气氛围下,以20℃/min的升温速率从室温升至900℃。
2 结果与讨论
2.1 催化剂表征
2.1.1 透射电子显微镜(TEM)
图 2为3%Pd/nano-SiO2催化剂还原以后的TEM照片。
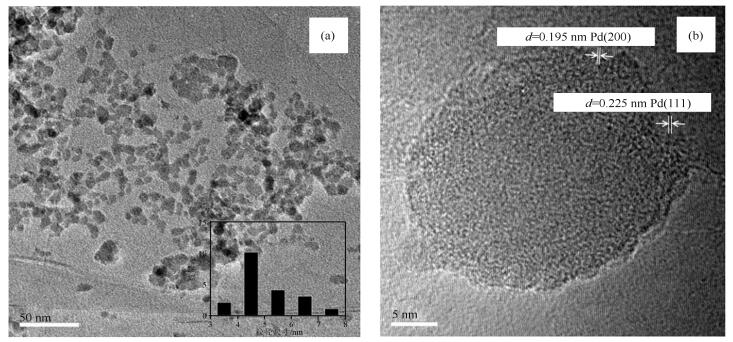 图2
Pd/nano-SiO2催化剂还原后的TEM照片
Figure2.
TEM image of Pd/nano-SiO2
图2
Pd/nano-SiO2催化剂还原后的TEM照片
Figure2.
TEM image of Pd/nano-SiO2
由图 2(a)可知,催化剂活性组分分布较为均匀,颗粒粒径较小,仅为5.1nm,这比使用PdCl2作为前驱体浸渍法制备的Pd/SiO2的颗粒粒径要小很多[30, 31],表明与普通SiO2相比,nano-SiO2作为载体对于活性组分有更好的分散作用。在图 2(b)中可以观察到Pd的晶格条纹,通过测量条纹间距发现分别对应于Pd的(111) 晶面和(200) 晶面,说明还原后的催化剂上出现了有催化活性的金属态活性组分。
2.1.2 热重分析
生物油中的含氧有机物受热可能会发生缩合积炭反应,因而对不同压力下200、250和300℃加氢反应后的催化剂进行热重分析,结果见表 1。计算得到的积炭量结果表明随着加氢温度升高,积炭反应受到了抑制。
 表 1
反应后催化剂的积炭
Table 1.
Coke deposition of spent catalysts
表 1
反应后催化剂的积炭
Table 1.
Coke deposition of spent catalysts
Conditions(℃/MPa) Coke/Catalyst(g/g) 200/2 0.073 250/2 0.063 300/2 0.055 200/3 0.075 250/3 0.061 300/3 0.057 200/4 0.071 250/4 0.059 300/4 0.055 2.2 温和加氢实验
2.2.1 反应物转化率
考察了模拟生物油与乙醇在200-300℃和2-4MPa下的反应活性,不同工况下的整体转化率见图 3,实验结果表明,反应温度和压力的升高都有利于模拟生物油的转化。由图 3(a)可知,在200℃下,增加压力至4MPa,整体转化率由80.1%提高到91.2%,这主要归因于各个反应物转化率的提高,其中, 环戊酮、愈创木酚、苯酚的转化率分别由39.8%、43.1%和52.5%提高至65.7%、50.4%和62.2%,而羟基丙酮的转化率也由87.2%提高到99.8%。由图 3(b)可知,在4MPa下,提高温度至300℃,整体转化率由91.2%继续提高至93.8%,其中, 愈创木酚、苯酚的转化率则显著增加到73.2%和83.0%。愈创木酚和苯酚在300℃时才具有较高的转化率,Grange等[32]也发现,由于不同化合物加氢反应的活化能不同,不同含氧官能团加氢的难易程度不同,愈创木酚需要达到300℃左右才能实现比较好的转化。此外,由于醛基反应活性较强,糠醛在200℃/2MPa下的转化率已经达到90.3%,当温度和压力提升后也实现了完全转化。乙酸的转化率在各个工况点均超过了98%。
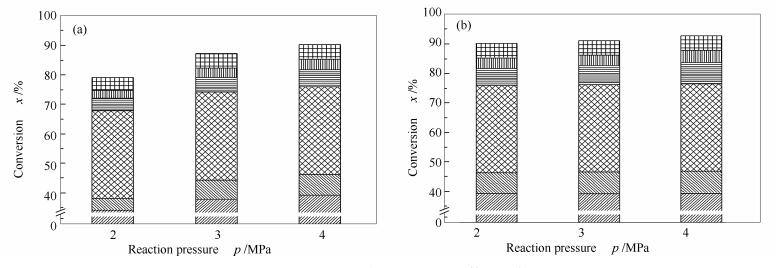 图3
不同工况下模拟生物油的整体转化率
Figure3.
Overall conversions of simulated bio-oil under different conditions
图3
不同工况下模拟生物油的整体转化率
Figure3.
Overall conversions of simulated bio-oil under different conditions
2.2.2 产物的选择性
温和加氢得到的液体产物为淡黄色透明不分层液体。不同温度和压力下的产物选择性见表 2。200℃时,各个压力下的液体产物选择性接近90%。当温度提高到250℃时,液体产物选择性均略有升高。相关研究表明,生物油中的酚类和醛类等含氧化合物受热可能发生缩合积炭反应[7, 8],从而导致积炭生成。前文提到,提高温度可以促进含氧化合物的转化,从而抑制其进一步缩合;而根据热重分析结果,4MPa下200、250和300℃时催化剂的积炭量分别为0.071、0.059和0.055g/g,其他压力下的积炭规律与之相同,表明随着加氢温度升高,积炭反应确实受到了抑制,更多的碳保留在液体产物中,这与250℃的液体产物选择性高于200℃的实验结果相符合。李其义等[33]在生物油的加氢脱氧研究中也发现,温度从200℃升高至240℃时,焦炭产率呈下降趋势。当温度继续提高到300℃时,促进了气体产物的生成,液体产物选择性因而略微下降。结合反应物转化率和液体产物选择性,250-300℃是比较合适的加氢温度范围。Elliott等[20]在乙酸和愈创木酚的加氢研究中也发现,Pd基催化剂更适合用于高温加氢,并在300℃下获得了更好的加氢效果。在2-4MPa的低压下,压力的变化对液体选择性的影响并不明显。
表 2
不同工况下产物的选择性
Table 2.
Product selectivities under different conditions
Conditions(℃/MPa) Product selectivity s/% liquid products CO CH4 C2H6 C3H8 C4H10 200/2 89.3 0 0 0 0 0.43 250/2 92.3 0 0.03 0.04 0.43 0.37 300/2 88.7 2.01 0.09 0.37 0.51 0.22 200/3 88.8 0 0 0 0 0.29 250/3 91.9 0 0.02 0.03 0.42 0.26 300/3 87.1 2.27 0.11 0.39 0.51 0.33 200/4 88.0 0 0 0 0 0.30 250/4 91.9 0 0 0.03 0.50 0.26 300/4 85.0 3.00 0.28 0.78 0.50 0.28 200℃时,各个压力下得到的气体产物只有少量丁烷,可能是糠醛加氢过程中开环加氢脱氧得到的气体产物[34]。250℃时,气体产物仍然很少,但与200℃相比,开始出现C1-3的气态烃类,表明温度提高后断键作用加强。当温度继续提高到300℃时,气体产物增加,CO开始生成并成为主要产物,表明此时脱羰基反应开始发生。产物中并未观测到CO2的生成,说明脱羧基反应强度较弱。在250℃以下,压力对气体产物选择性的影响不明显,当温度达到300℃时,随着压力的提高,气体产物选择性有明显提高,这可能是由于在较高的温度和压力共同作用下,促进了含氧化合物的加氢裂解和脱氧反应,生成了较多的CO和少量的气态烃类。
2.2.3 液体产物分析
结合上述实验结果,初步确定250-300℃和4MPa为比较合适的工况。为了进一步研究温和加氢对模化物有效氢碳比的影响并探索最佳的加氢工况,对不同工况下的液体产物(包括溶剂乙醇和未转化的反应物)进行了元素分析,并依据公式(1) 计算相应的有效氢碳比,所得结果见图 4。
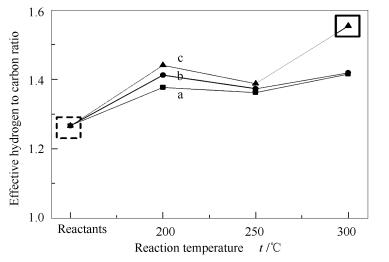 图4
不同工况下液体产物的有效氢碳比
Figure4.
(H/C)eff of liquid products under different conditions
图4
不同工况下液体产物的有效氢碳比
Figure4.
(H/C)eff of liquid products under different conditions
根据反应原料中模拟生物油和乙醇的比例,依据公式(1) 计算得到反应物的有效氢碳比为1.266。经过各个工况的加氢反应后,产物的有效氢碳比均超过了1.350,表明温和加氢对于模化物有效氢碳比的提高有明显的促进作用。同时,温度和压力对于产物有效氢碳比有明显的影响。在200℃/2MPa的工况下,产物有效氢碳比为1.376。将压力提高至3和4MPa,产物有效氢碳比分别提高到1.412和1.441。250和300℃时的结果也与此相似。值得注意的是,随着反应温度的升高,产物有效氢碳比呈先略微下降后升高的趋势。结合热重分析和元素分析发现,与200℃相比,250℃时的积炭量在各压力下均有明显下降,表明积炭反应受到抑制,使得液体产物中碳含量增加,同时脱氧效率仍然较低,因此, 根据公式(1) 有效氢碳比反而略有减少。当温度升高到300℃时,Pd/nano-SiO2活性增强,更多的氢进入到液体产物中,同时部分氧以CO的形式脱除,使得该温度下有效氢碳比较大。在300℃/4MPa的工况下,产物有效氢碳比达到最大值,为1.554,因此, 该工况下加氢产物的反应活性明显提高,最适合用于催化裂化[12]。
结合反应物转化率、液体产物选择性、气体产物组成及液体产物有效氢碳比,确定本研究中温和加氢的最佳工况为300℃/4MPa。利用GC-MS对该工况下的液体产物组成进行分析,得到的谱图见图 5(除了部分未转化的环戊酮、愈创木酚和苯酚,主要反应产物用箭头进行标示)。
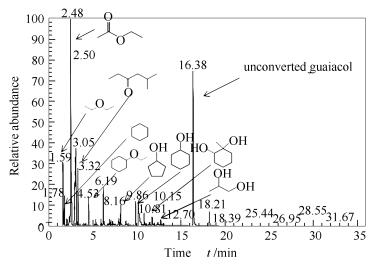 图5
300℃/4MPa下液体产物的GC-MS谱图
Figure5.
GC-MS spectrum of liquid products (300℃/4MPa)
图5
300℃/4MPa下液体产物的GC-MS谱图
Figure5.
GC-MS spectrum of liquid products (300℃/4MPa)
根据化合物的结构和官能团对液体产物进行归类(未转化的反应物不计入相对含量的计算),发现其主要包含酯类(44.1%)、醇类(19.0%)、醚类(16.9%)、酮类(11.9%)、烃类(4.2%)、醛类(1.5%)、酸类(1.4%)和呋喃类(1.0%)。酯类中乙酸乙酯含量最高,为40.1%,是乙酸和乙醇发生酯化反应的产物,这也表明乙酸主要发生的是酯化反应而非脱羧基反应,与气体产物中未观测到CO2生成的实验结果相符,同时部分乙酸也可能直接加氢生成少量乙醇[20]。醚类中的二乙醚(7.7%)主要来源于乙醇的分子间脱水反应,其含量尽管位列第二,但明显低于乙酸乙酯。醇类中绝大多数为反应物加氢生成的对应产物,主要包含环己醇(5.6%)、环戊醇(4.3%)、环己二醇(2.4%)和1, 2-丙二醇(1.9%)。其中, 环戊醇、1, 2-丙二醇分别为环戊酮和羟基丙酮加氢的产物,环己醇、环己二醇为苯酚和愈创木酚加氢的主要产物。由于乙酸的存在,部分1, 2-丙二醇与乙酸发生酯化反应,生成了少量1, 2-丙二醇二乙酯、1, 2-丙二醇-2-乙酯,因此, 产物中1, 2丙二醇含量较少。反应物中的糠醛除了转化为气体产物丁烷外,还生成了少量糠醇和四氢呋喃的同族物。酮类主要是苯酚和愈创木酚加氢产生的中间产物环己酮及其同族物[35, 36]以及开环产物,如5-甲基-3-己酮(5.5%)。烃类主要为环己烷(3.7%),是环己醇进一步加氢脱氧的产物。反应物的主要生成路径见图 6。
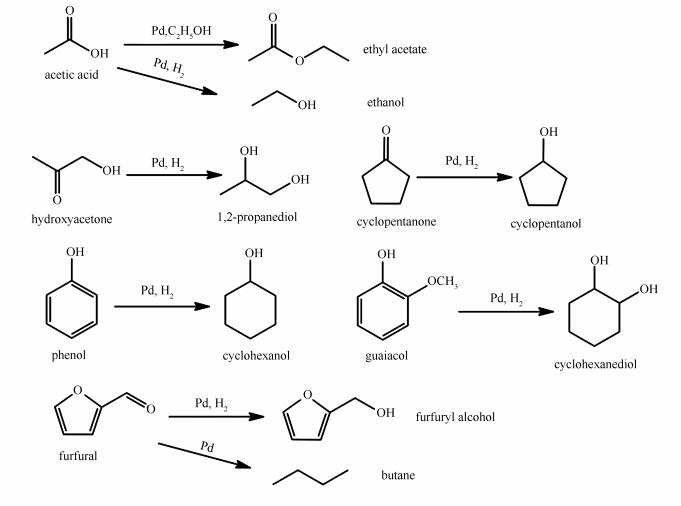 图6
反应物的主要反应路径示意图
Figure6.
Main reaction pathway of reactants
图6
反应物的主要反应路径示意图
Figure6.
Main reaction pathway of reactants
以上结果表明,液体产物中具有较多的酯类、醇类、醚类和酮类,而几乎不含酸类和酚类。过去研究者曾对族类化合物反应活性进行了考察,Gayubo等[7, 8]发现,羟基相比于羰基和羧基具有更高的反应活性,因而醇类比酸、酮类更容易转化;酮类向烃类的转化效率要略低于醇类而要高于酸类、醛类和酚类。Adjaye等[37]在生物油模化物裂化反应机理的研究中发现,酯类在HZSM-5催化作用下的裂化效率明显高于酸类,因此, 液体产物组成中较多的乙酸乙酯有利于后续的催化裂化。Ni等[38]在醇醚类芳构化的研究中发现,二乙醚向苯、甲苯和二甲苯转化的选择性达到了56.8%,30.6%转化为C1-4的气态烃类,表明二乙醚也具有较好的裂化特性。因此,综合族类化合物的反应活性研究,300℃/4MPa的产物分布更加适合开展裂化工况的研究,与之前的分析相符合。
3 结论
本研究主要通过温和加氢手段实现了模拟生物油组成的明显优化和有效氢碳比的明显提高。首先使用3%Pd/nano-SiO2催化剂在不同工况下开展温和加氢研究,发现300℃/4MPa是最佳的加氢工况,该条件下反应物除苯酚和愈创木酚外几乎完全转化,液体产物选择性为85.0%,有效氢碳比从初始反应物的1.266明显提升至1.554,液体产物组成得到优化,酚类和酸类的含量明显下降,反应活性明显改善,有利于后续的催化裂化反应。
-
-
[1]
CZERNIK S, BRIDGWATER A V. Overview of applications of biomass fast pyrolysis oil[J]. Energy Fuels, 2004, 18(2): 590-598. doi: 10.1021/ef034067u
-
[2]
王琦, 刘倩, 贺博, 王树荣, 骆仲泱, 岑可法. 流化床生物质快速热解制取生物油试验研究[J]. 工程热物理学报, 2008,29,(5): 885-888. WANG Qi, LIU Qian, HE Bo, WANG Shu-rong, LUO Zhong-yang, CEN Ke-fa. Experimental research on biomass flash pyrolysis for bio-oil in a fluidized bed reactor[J]. J Eng Thermophys, 2008, 29(5): 885-888.
-
[3]
ZHANG Q, CHANG J, WANG T J, XU Y. Review of biomass pyrolysis oil properties and upgrading research[J]. Energy Convers Manage, 2007, 48(1): 87-92. doi: 10.1016/j.enconman.2006.05.010
-
[4]
XIU S, SHAHBAZI A. Bio-oil production and upgrading research:A review[J]. Renewable Sustainable Energy Rev, 2012, 16(7): 4406-4414. doi: 10.1016/j.rser.2012.04.028
-
[5]
郭晓亚, 颜涌捷. 生物质快速裂解油的催化裂解精制[J]. 化学反应工程与工艺, 2005,21,(3): 227-233. GUO Xiao-ya, YAN Yong-jie. Catalytic cracking of biomass fast pyrolysis oil[J]. Chem React Eng Technol, 2005, 21(3): 227-233.
-
[6]
陈娇娇, 陈冠益, 马文超, 马隆龙, 王铁军, 张琦, 吕微. 生物油模型化合物催化裂化制备芳香烃的实验研究[J]. 燃料化学学报, 2013,41,(2): 183-188. CHEN Jiao-jiao, CHEN Guan-yi, MA Wen-chao, MA Long-long, WANG Tie-jun, ZHANG Qi, LÜ Wei. Experimental study of aromatics production from catalytic cracking of bio-oil model compounds[J]. J Fuel Chem Technol, 2013, 41(2): 183-188.
-
[7]
GAYUBO A G, AGUAYO A T, ATUTXA A, AGUADO R, BILBAO J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols[J]. Ind Eng Chem Res, 2004, 43(11): 2610-2618. doi: 10.1021/ie030791o
-
[8]
GAYUBO A G, AGUAYO A T, ATUTXA A, AGUADO R, OLAZAR M, BILBAO J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite.Ⅱ. Aldehydes, ketones, and acids[J]. Ind Eng Chem Res, 2004, 43(11): 2619-2626. doi: 10.1021/ie030792g
-
[9]
CARLSON T R, JAE J, LIN Y C, TOMPSETT G A, HUBER G W. Catalytic fast pyrolysis of glucose with HZSM-5:The combined homogeneous and heterogeneous reactions[J]. J Catal, 2010, 270(1): 110-124. doi: 10.1016/j.jcat.2009.12.013
-
[10]
王誉蓉, 王树荣, 王相宇, 郭祚刚. 不同蒸馏压力下的生物油分子蒸馏分离特性研究[J]. 燃料化学学报, 2013,41,(2): 177-182. WANG Yu-rong, WANG Shu-rong, WANG Xiang-yu, GUO Zuo-gang. Molecular distillation separation characteristic of bio-oil under different pressures[J]. J Fuel Chem Technol, 2013, 41(2): 177-182.
-
[11]
GUO Z G, WANG S R, GU Y L, XU G H, LI X, LUO Z Y. Separation characteristics of biomass pyrolysis oil in molecular distillation[J]. Sep Purif Technol, 2010, 76(1): 52-57. doi: 10.1016/j.seppur.2010.09.019
-
[12]
MENTZEL U V, HOLM M S. Utilization of biomass:Conversion of model compounds to hydrocarbons over zeolite H-ZSM-5[J]. Appl Catal A, 2011, 396(1/2): 59-67.
-
[13]
WANG S R, CAI Q J, WANG X Y, ZHANG L, WANG Y R, LUO Z Y. Biogasoline production from the Co-cracking of the distilled fraction of bio-oil and ethanol[J]. Energy Fuels, 2014, 28(1): 115-122. doi: 10.1021/ef4012615
-
[14]
WANG S R, CAI Q J, WANG X Y, ZHANG L, WANG Y R, LUO Z Y. Biogasoline production by co-cracking of model compound mixture of bio-oil and ethanol over HSZM-5[J]. Chin J Catal, 2014, 35(5): 709-722. doi: 10.1016/S1872-2067(14)60046-2
-
[15]
WANG S R, CAI Q J, CHEN J H, ZHANG L, WANG X Y, YU C J. Green aromatic hydrocarbon production from cocracking of a bio-oil model compound mixture and ethanol over Ga2O3/HZSM-5[J]. Ind Eng Chem Res, 2014, 53(36): 13935-13944. doi: 10.1021/ie5024029
-
[16]
WANG S R, CAI Q J, CHEN J H, ZHANG L, ZHU L J, LUO Z Y. Co-cracking of bio-oil model compound mixtures and ethanol over different metal oxide-modified HZSM-5 catalysts[J]. Fuel, 2015, 160: 534-543. doi: 10.1016/j.fuel.2015.08.011
-
[17]
VENDERBOSCH R H, ARDIYANTI A R, WILDSCHUT J, OASMAA A, HEERES H J. Stabilization of biomass-derived pyrolysis oils[J]. J Chem Technol Biotechnol, 2010, 85(5): 674-686. doi: 10.1002/jctb.v85:5
-
[18]
WILDSCHUT J, MAHFUD F H, VENDERBOSCH R H, HEERES H J. Hydrotreatment of fast pyrolysis oil using heterogeneous noble-metal catalysts[J]. Ind Eng Chem Res, 2009, 48(23): 10324-10334. doi: 10.1021/ie9006003
-
[19]
ARDIYANTI A R, GUTIERREZ A, HONKELA M L, KRAUSE A O I, HEERES H J. Hydrotreatment of wood-based pyrolysis oil using zirconia-supported mono-and bimetallic (Pt, Pd, Rh) catalysts[J]. Appl Catal A, 2011, 407(1/2): 56-66.
-
[20]
ELLIOTT D C, HART T R. Catalytic hydroprocessing of chemical models for bio-oil[J]. Energy Fuels, 2009, 23(2): 631-637. doi: 10.1021/ef8007773
-
[21]
CHEN J H, CAI Q J, LU L, LENG F R, WANG S R. Upgrading of the acid-rich fraction of bio-oil by catalytic hydrogenation-esterification[J]. ACS Sustainable Chem Eng, 2016, 5(1): 1073-1081.
-
[22]
姚燕, 王树荣, 骆仲泱, 岑可法. 生物油轻质馏分加氢试验研究[J]. 工程热物理学报, 2008,29,(4): 715-719. YAO Yan, WANG Shu-rong, LUO Zhong-yang, CEN Ke-fa. Experimental research on catalytic hydrogenation of light fraction of bio-oil[J]. J Eng Thermophys, 2008, 29(4): 715-719.
-
[23]
YU W J, TANG Y, MO L Y, CHEN P, LOU H, ZHENG X M. One-step hydrogenation-esterification of furfural and acetic acid over bifunctional Pd catalysts for bio-oil upgrading[J]. Bioresour Technol, 2011, 102(17): 8241-8246. doi: 10.1016/j.biortech.2011.06.015
-
[24]
YU WJ, TANG Y, MO L Y, CHEN P, LOU H, ZHENG X M. Bifunctional Pd/Al-SBA-15 catalyzed one-step hydrogenation-esterification of furfural and acetic acid:A model reaction for catalytic upgrading of bio-oil[J]. Catal Commun, 2011, 13(1): 35-39. doi: 10.1016/j.catcom.2011.06.004
-
[25]
仲卫成, 郭庆杰, 王许云, 张亮. 小球藻热裂解油催化加氢精制研究[J]. 燃料化学学报, 2013,41,(5): 571-578. ZHONG Wei-cheng, GUO Qing-jie, WANG Xu-yun, ZHANG Liang. Catalytic hydroprocessing of fast pyrolysis bio-oil from Chlorella[J]. J Fuel Chem Technol, 2013, 41(5): 571-578.
-
[26]
ELLIOTT D C. Historical Developments in Hydroprocessing Bio-oils[J]. Energy Fuels, 2007, 21(3): 1792-1815. doi: 10.1021/ef070044u
-
[27]
魏宏鸽, 仲兆平, 李睿, 靳立维. 固体催化剂下生物油模型化合物的催化加氢研究[J]. 可再生能源, 2010,28,(1): 52-62. WEI Hong-ge, ZHONG Zhao-ping, LI Rui, JIN Li-wei. Research on solid catalysts base hydrogenation for bio-oil model compounds[J]. Renewable Energy Resour, 2010, 28(1): 52-62.
-
[28]
ZHAO H Y, LI D, BUI P, OYAMA S T. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing Catalysts[J]. Appl Catal A, 2011, 391(1/2): 305-310.
-
[29]
VISPUTE T P, ZHANG H Y, SANNA A, XIAO R, HUBER G W. Renewable chemical commodity feedstocks from integrated catalytic processing of pyrolysis oils[J]. Science, 2010, 330(6008): 1222-1227. doi: 10.1126/science.1194218
-
[30]
BECK A, HORVATH A, SZUCS A, SCHAY Z, HORVATH Z E, ZSOLDOS Z, DEKANY I, GUCZI L. Pd nanoparticles prepared by "controlled colloidal synthesis" in solid/liquid interfacial layer on silica. I. Particle size regulation by reduction time[J]. Catal Lett, 2000, 65(1/3): 33-42. doi: 10.1023/A:1019048701152
-
[31]
LI B, WENG W Z, ZHANG Q, WANG Z W, WAN H L. Sinter-resistant Pd/SiO2 nanocatalyst prepared by impregnation method[J]. ChemCatChem, 2011, 3(8): 1277-1280. doi: 10.1002/cctc.201100043
-
[32]
GRANGE P, LAURENT E, MAGGI R, CENTENO A, DELMON B. Hydrotreatment of pyrolysis oils from biomass:Reactivity of the various categories of oxygenated compounds and preliminary techno-economical study[J]. Catal Today, 1996, 29(1/4): 297-301.
-
[33]
李其义, 万磊, 张素平, 许庆利, 颜涌捷. 生物油低温加氢脱氧的研究[J]. 石油化工, 2011,40,(9): 954-958. LI Qi-yi, WAN Lei, ZHANG Su-ping, XU Qing-li, YAN Yong-jie. Hydrodeoxygenation of bio-oil under mild conditions[J]. Petrochem Technol, 2011, 40(9): 954-958.
-
[34]
KREUZER K, KRAMER R. Support effects in the hydrogenolysis of tetrahydrofuran on platinum catalysts[J]. J Catal, 1997, 167(2): 391-399. doi: 10.1006/jcat.1997.1608
-
[35]
ZHAO C, HE J Y, LEMONIDOU A A, LI X B, LERCHER J A. Aqueous-phase hydrodeoxygenation of bio-derived phenols to cycloalkanes[J]. J Catal, 2011, 280(1): 8-16. doi: 10.1016/j.jcat.2011.02.001
-
[36]
SHAFAGHAT H, REZAEI P S, DAUD W M A W. Catalytic hydrogenation of phenol, cresol and guaiacol over physically mixed catalysts of Pd/C and zeolite solid acids[J]. RSC Adv, 2015, 5(43): 33990-33998. doi: 10.1039/C5RA00367A
-
[37]
ADJAYE J D, BAKHSHI N N. Catalytic conversion of a biomass-derived oil to fuels and chemicals Ⅰ:Model compound studies and reaction pathways[J]. Biomass Bioenergy, 1995, 8(3): 131-149. doi: 10.1016/0961-9534(95)00018-3
-
[38]
NI Y M, PENG W Y, SUN A M, MO W L, HU J L, LI T, LI G X. High selective and stable performance of catalytic aromatization of alcohols and ethers over La/Zn/HZSM-5 catalysts[J]. J Ind Eng Chem, 2010, 16(4): 503-505. doi: 10.1016/j.jiec.2010.03.011
-
[1]
-

图 1 生物油催化加氢固定床反应系统示意图
Figure 1 Fixed-bed reaction system for bio-oil catalytic hydrogenation

图 3 不同工况下模拟生物油的整体转化率
Figure 3 Overall conversions of simulated bio-oil under different conditions
(a): the effect of reaction pressure (200℃); (b): the effect of reaction temperature (4MPa)

图 4 不同工况下液体产物的有效氢碳比
Figure 4 (H/C)eff of liquid products under different conditions
a: 2MPa; b: 3MPa; c: 4MPa
表 1 反应后催化剂的积炭
Table 1. Coke deposition of spent catalysts
Conditions(℃/MPa) Coke/Catalyst(g/g) 200/2 0.073 250/2 0.063 300/2 0.055 200/3 0.075 250/3 0.061 300/3 0.057 200/4 0.071 250/4 0.059 300/4 0.055  下载: 导出CSV
下载: 导出CSV
表 2 不同工况下产物的选择性
Table 2. Product selectivities under different conditions
Conditions(℃/MPa) Product selectivity s/% liquid products CO CH4 C2H6 C3H8 C4H10 200/2 89.3 0 0 0 0 0.43 250/2 92.3 0 0.03 0.04 0.43 0.37 300/2 88.7 2.01 0.09 0.37 0.51 0.22 200/3 88.8 0 0 0 0 0.29 250/3 91.9 0 0.02 0.03 0.42 0.26 300/3 87.1 2.27 0.11 0.39 0.51 0.33 200/4 88.0 0 0 0 0 0.30 250/4 91.9 0 0 0.03 0.50 0.26 300/4 85.0 3.00 0.28 0.78 0.50 0.28
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 2216
- HTML全文浏览量: 289
 下载:
下载:
