 图 图式1
文献报道的拉帕替尼的合成路线
Figure 图式1.
The reported synthetic routes for lapatinib in literatures
图 图式1
文献报道的拉帕替尼的合成路线
Figure 图式1.
The reported synthetic routes for lapatinib in literatures
引用本文:
郭卿, 李雁武, 任学状, 袁建勇. 拉帕替尼新中间体的合成[J]. 化学通报,
2017, 80(4): 392-395.

Citation: Guo Qing, Li Yanwu, Ren Xuezhuang, Yuan Jianyong. Synthesis of a Novel Intermediate of Lapatinib[J]. Chemistry, 2017, 80(4): 392-395.
Citation: Guo Qing, Li Yanwu, Ren Xuezhuang, Yuan Jianyong. Synthesis of a Novel Intermediate of Lapatinib[J]. Chemistry, 2017, 80(4): 392-395.
拉帕替尼新中间体的合成
English
Synthesis of a Novel Intermediate of Lapatinib
Abstract:
A Schiff base was synthesized from commercially available 5-bromo-2-furaldehyde and 2-(methylsulfonyl)ethaneamine in the nucleophilic additional reaction. A novel intermediate of lapatinib was synthesized from the Schiff base by hydrogenation reduction, amino protection, borylation reaction. The overall yield is 66.32%.
-
Key words:
- Lapatinib
- / Organomagnesium complex
- / Synthesis
-
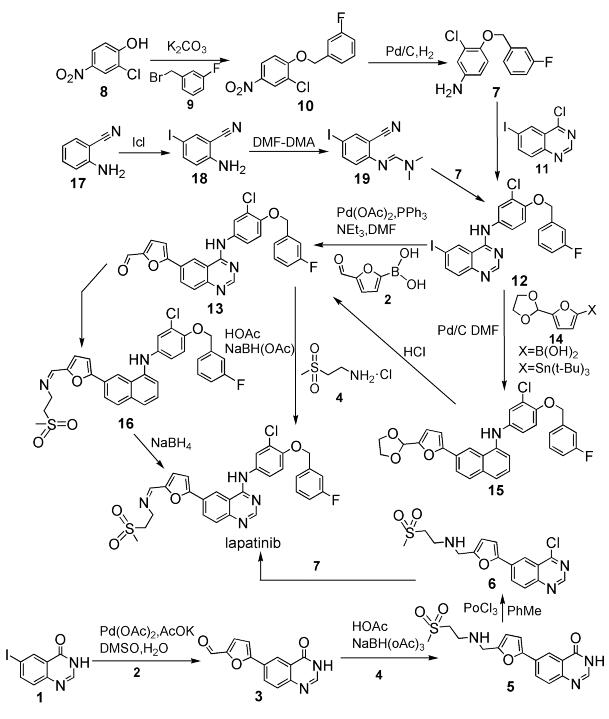
拉帕替尼(lapatinib),化学名N-(3-氯-4-((3-氟苯基)甲氧基)苯基)-6-(5-(((2-(甲磺酰基)乙基)氨基)甲基)-呋喃2-基)-4-喹唑啉胺二对甲苯磺酸盐,分子式:C29H26ClFN4O4S·2C7H8O3S,分子量:925.46,是一种口服的小分子可逆性EGFR和HER2双受体阻断剂。实验研究中发现,拉帕替尼对HER2阳性乳腺癌细胞,以及曲妥单抗空调乳腺癌细胞有很好的抑制作用[1~3]。在2005年,拉帕替尼因对ErbB2过度表达或者前期治疗失败的晚期或转移性乳腺癌患者的良好治疗作用而被FDA列入了快速审批的名单中[4]。因其特殊疗效,拉帕提尼成为了备受关注的抗癌药品,同时,其几种合成路线(见图式 1) 也被开发出来。
图 图式1
文献报道的拉帕替尼的合成路线
Figure 图式1.
The reported synthetic routes for lapatinib in literatures
报道的合成路线之一是由6-碘喹唑啉-4(3H)-酮(1)与5-甲酰基呋喃-2-硼酸(2)经过Suzuki偶联反应得到5-(4-氧代-3, 4-二氢喹唑啉-6-基)呋喃-2-甲醛(3),而后3与2-甲磺酰基乙胺盐酸盐(4)经过还原氨化得到5,5经过氯代得到6,最后6与3-氯-4-(3-氟苄氧基)苯胺(7)反应得到目标化合物拉帕替尼。该合成路线的优势在于反应步骤少,但是路线中用到了三氯氧磷这种易挥发的有毒刺激性液体,不利于环保。报道的合成路线二是由以2-氯4-硝基苯酚(8)与3-氟溴苄(9)反应生成3-氯-4-(3-氟苄氧基)硝基苯(10),而后10经过还原氢化得到7,7与6-碘喹唑啉-4(3H)-酮(11)缩合制得N-[3-氯-4-(3-氟苄氧基)苯基]-6-碘-4-喹唑啉胺(12),12与2经过Suzuki偶联反应得到(13),13与4反应制得最终产物拉帕替尼。或者由中间体12与5-硼酸基-2-呋喃甲醛缩乙二醇或5-(三叔丁基锡)-2-呋喃甲醛缩乙二醇(14)经钯炭催化偶联得15,而后15经过盐酸酸化脱去保护基得到13,13与4反应制得16,而后经过氢化还原得到终产物拉帕替尼。该路线操作繁琐,且其中要用到三苯基膦这种后处理不易除去的化合物,间接影响了产品的纯度。合成路线三是由2-氨基苄腈(17)经过碘代反应生成2-氨基-5-碘苄腈(18),而后18与DMF-DMA反应得到19,19与7反应得到中间体12,然后再经过两步反应得到最终产物拉帕替尼。该条反应路线在初始碘代环节中使用氯化碘这种高毒无机物,不利于环保,且整个路线步骤长、操作繁琐,不利于大规模生产[5, 6]。
考虑到以上三条路线的不足,本文采用的合成方法是通过5-溴糠醛(20)与2-甲磺酰基乙胺(4)反应生成21,而后21经过氢化还原得到化合物22,22用溴苄进行氨基保护后与硼酸酯反应制得中间体24[7],该中间体是一种未见报道的新化合物,具有进一步研究价值。其可以通过与12进行Suzuki偶联反应得到25,25经过选择性脱苄得到最终产物拉帕替尼(见图式 2),本文重点讨论新中间体的合成。
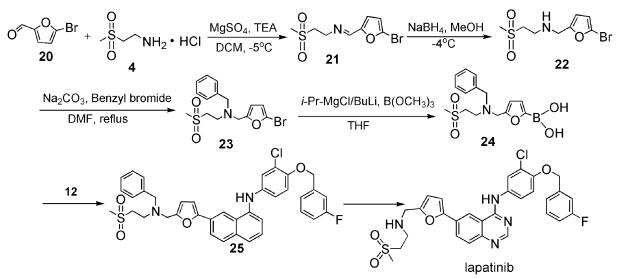 图 图式2
本文的拉帕替尼的合成路线
Figure 图式2.
A new synthetic route for the preparation of lapatinib
图 图式2
本文的拉帕替尼的合成路线
Figure 图式2.
A new synthetic route for the preparation of lapatinib
1 实验部分
1.1 仪器与试剂
DLSB-5/20型低温恒温反应浴(郑州长城科工贸有限公司);SHB-IIIS型循环水多用真空泵(郑州长城科工贸有限公司);CS101-1EBW型电热鼓风干燥箱(重庆万达仪器有限公司);ZNCL-GS型磁力搅拌器(河南爱博特科技发展有限公司);DF-101S型集热式恒温加热磁力搅拌器(重庆东悦仪器有限公司);旋转蒸发仪(Heidolph);ACO型电磁式空气泵(森森集团股份有限公司);DZF-6020型真空干燥箱(上海博远实业有限公司);2034型真空隔膜泵(WELCH);ZF-IⅠ型三用紫外分析仪(上海顾村电光仪器厂);KF-188A型电吹机(广州华能达电气有限公司);D60-2F型电动搅拌机(杭州仪表电机有限公司);YRT-3型熔点仪(南京旭析仪器有限公司);400MHz核磁共振谱仪(德国Bruker公司)。
2-(甲基砜)乙胺盐酸盐(95%,百灵威科技有限公司);5-溴糠醛(98%,萨恩化学技术(上海)有限公司);无水硫酸镁(AR,成都市科龙化工试剂厂);硼氢化钠(98%,阿拉丁试剂上海有限公司);无水碳酸钠(AR,重庆川东化工有限公司);溴苄(AR,阿拉丁试剂上海有限公司);异丙基氯化镁(2.0mol/L in THF,萨恩化学技术(上海)有限公司);正丁基锂(1.6mol/L in hexanes,阿达玛斯试剂有限公司); 硼酸三甲酯(98%,萨恩化学技术(上海)有限公司);四氢呋喃(AR,上海晶纯试剂有限公司);二氯甲烷(AR,成都市科龙化工试剂厂);三乙胺(AR,重庆川东化工有限公司);甲醇(AR,重庆川东化工有限公司);乙酸乙酯(AR,成都市科龙化工试剂厂)。
1.2 实验部分
1.2.4 5-((苄基(2-(甲基磺酰基)乙基)氨基)甲基)呋喃-2-基硼酸(24)的合成
氮气保护下,在三口瓶中加入50mL干燥的THF,将三口瓶移入0℃的低温冷却槽中,而后分别滴加3.75mL(7.50mmol)异丙基氯化镁和9.73mL(15mmol)丁基锂,搅拌15min,降温至-20℃,滴加5.4g (15mmol)上述合成的23的30mL THF溶液,搅拌反应30min,而后滴加8.96g(60mmol)硼酸三甲酯。滴加完毕后逐渐将内温升至0℃,保持该温度搅拌反应2h。反应结束后,混合液用醋酸猝灭,加入少许水稀释,水层用乙酸乙酯(50mL×3) 萃取,有机相用饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩得半固体状产品,粗产品经柱层析(洗脱液,二氯甲烷/乙酸乙酯=2/1) 提纯得到3.71g化合物24(产率73.39%)。1H NMR (400MHz,DMSO-d6) δ:7.31~7.13 (m,5H),6.12 (s,1H),3.76 (s,2H),3.60 (s,2H),3.15 (d,J=3.9Hz,3H),3.03 (s,1H),2.80 (s,3H),1.55 (s,2H)。
1.2.3 N-苄基-N-((5-溴呋喃-2-基)甲基)-2-(甲基磺酰基)乙胺(23)的合成
在反应瓶中加入7.00g(25.00mmol)上述合成的22、5.29g(50.00mmol)碳酸钠和50mL DMF,而后缓慢加入4.27g(30.00mmol)溴苄,升温至140℃,搅拌反应3h。反应液冷却后,加入乙酸乙酯,用水(30mL×3) 洗涤,合并有机相,而后用饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压浓缩得到浅黄色固体8.91g (产率96.53%)。1H NMR (400MHz,CDCl3)δ:7.39~7.29 (m,5H),6.28 (d,J=3.2Hz,1H),6.22 (d,J=3.2Hz,1H),3.67 (d,J=9.9Hz,4H),3.08 (d,J=1.9Hz,4H),2.98 (s,3H)。
1.2.1 (E)-N-((5-溴呋喃-2-基)亚甲基)-2-(甲基磺酰基)乙胺(21) 的合成
在三口瓶中加入5.00g (28.56mmol)20和5.01g(31.38mmol)4,而后加入3.42g(28.50mmol)无水硫酸镁和干燥的75mL二氯甲烷,在低温冷却槽中保持-5℃下搅拌,全程氮气保护。向该混合物中缓慢滴加三乙胺(4.75g,46.90mmol),继续于-5℃下反应2h。反应结束后减压抽滤,除去硫酸镁,滤液用饱和氯化钠溶液洗涤,分液,有机相用无水硫酸钠干燥,最后减压浓缩得到浅黄色固体7.50g(产率94.25%)。1H NMR (400 MHz,CDCl3) δ:7.38 (s,1H),7.08 (d,J=15.0 Hz,1H),6.80 (d,J=15.0Hz,1H),3.89 (t,J=16.0Hz,2H),2.90 (t,J=15.9Hz,2H),2.80 (s,3H)。
1.2.2 N-((5-溴呋喃-2-基)甲基)-2-(甲基磺酰基)乙胺(22)的合成
在单口瓶中加入7.54g(26.9mmol)上述合成的21,加入150mL无水甲醇使原料溶解,在低温冷却槽中保持-4℃下搅拌,加入4.07g(107.6mmol)硼氢化钠,加样后,将反应液内温升至室温并继续搅拌1h。反应结束后将混合物降温至0℃,逐滴滴入饱和碳酸氢钠溶液60mL,而后将反应中的固体减压滤除,进一步用二氯甲烷(50mL×3) 洗涤滤饼,合并有机相,用饱和氯化钠溶液洗,分液,有机相用无水硫酸钠干燥,最后减压浓缩得到产品7.5g(产率99.33%)。1H NMR (400MHz,CDCl3)δ:6.24 (d,J=3.2Hz,1H),6.19 (d,J=3.2Hz,1H),3.77 (s,2H),3.14 (d,J=2.5Hz,4H),3.00 (s,3H),1.84 (s,1H)。
2 结果和讨论
我们对拉帕提尼的合成路线进行了详细的前期调研工作,最终选择5-溴糠醛(20)与2-甲磺酰基乙胺盐酸盐(4)作为起始原料,通过亲和取代反应生成化合物21[8, 9],而后,21经过氢化还原的到化合物22[10],22用溴苄做氨基保护得到化合物23[11, 12],23经过取代反应制得中间体24,总收率为66.32%。其中,化合物22可以通过一锅法进行连续反应,只需在合成化合物21后将反应溶剂二氯甲烷减压蒸干,得到的混合物便可直接进入下一步合成N-((5-溴呋喃-2-基)甲基)-2-(甲基磺酰基)乙胺(22),同时,在化合物22的反应后处理中,我们选用丙酮作溶剂在低温下结晶纯化,可得到高纯度的产物,该法不需要使用柱层析纯化,适合工业化生产。在制备化合物24时,采用了i-PrMg/n-BuLi这种复合物在非低温条件下制备硼化物,该方法反应条件温和,仅需在-10℃条件下就能进行,便于工业化操作,且该方法中所形成的呋喃镁复合物适用于多种亲电反应,具有高收率。
3 结论
本路线选择5-溴糠醛与2-甲磺酰基乙胺盐酸盐作为起始原料通过四步反应得到目标中间体5-((苄基(2-(甲基磺酰基)乙基)氨基)甲基)呋喃-2-基硼酸。该合成方法方便实用,具有原料价廉易得、反应条件温和、收率高、反应可控等优点,并降低了生产成本以及设备要求,有良好的工业化应用前景。
-
-
[1]
B Boyd, J Bozzo, J Castañer. Drug. Future, 2005, 30(12): 1225~1239.
-
[2]
S C Robertson, J Tynan, D J Donoghue. Trends Genet., 2000, 16: 368. http://www.researchgate.net/publication/12415607_RTK_mutations_and_human_syndromes_when_good_receptors_turn_bad
-
[3]
P van der Geer, T Hunter, R A Lindberg. Ann. Rev. Cell Biol., 1994, 10: 251~337. http://www.researchgate.net/profile/Tony_Hunter/publication/15311407_Receptor_protein-kinases_and_their_signal_transduction_pathways/links/02bfe513e442f17286000000.pdf
-
[4]
耿敬坤, 赵桂森. 中国医药工业杂志, 2012, 43(9): 796~798. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zhou201209027&dbname=CJFD&dbcode=CJFQ
-
[5]
Y F Chen, J P Henschke, Y L Liu et al. USP: 8563719, 2013. http://www.freepatentsonline.com/8563719.html
-
[6]
K G Petrov, Y M Zhang, M Carter et al. Bioorg. Med. Chem. Lett., 2006, 16(17):4686~4691. http://www.researchgate.net/publication/7007122_Optimization_and_SAR_for_dual_ErbB-1ErbB-2_tyrosine_kinase_inhibition_in_the_6-furanylquinazoline_series
-
[7]
K Satoshi, A Atsushi, A Takehiko et al. Tetrahed. Lett., 2006, 47: 1877~1879.
-
[8]
I Zubkov Fedor, V Nikitina Eugenia, R Galeev Timur et al. Tetrahedron, 2014, 70, (8):1659~1690. http://www.researchgate.net/publication/260033288_General_synthetic_approach_towards_annelated_3a6-epoxyisoindoles_by_tandem_acylationIMDAF_reaction_of_furylazaheterocycles._Scope_and_limitations
-
[9]
F I Zubkov, I K Airiyan, A A Dzyubenko et al. J. Heterocycl. Chem., 2010, 47 (2): 400~414.
-
[10]
C J Cooper, M D Jones, S K Brayshaw et al. Dalton Transac., 2011, 40(14), 3677~3682.
-
[11]
H Naoya, K Hiroyuki, O Masahiro et al. J. Organomet. Chem., 2011, 696(23), 3745~3749. http://www.researchgate.net/publication/251479001_Effective_synthesis_of_99mTc_tricarbonyl_complexes_by_microwave_heating?ev=auth_pub
-
[12]
X D Liu, A Patron, C Tachdjian et al. USP: 20090274632.
-
[1]
-

图式1 文献报道的拉帕替尼的合成路线
Scheme 1 The reported synthetic routes for lapatinib in literatures
-


 下载:
下载:

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 16
- 文章访问数: 2753
- HTML全文浏览量: 684
 下载:
下载:
